Advanced Simulation, Made Accessible
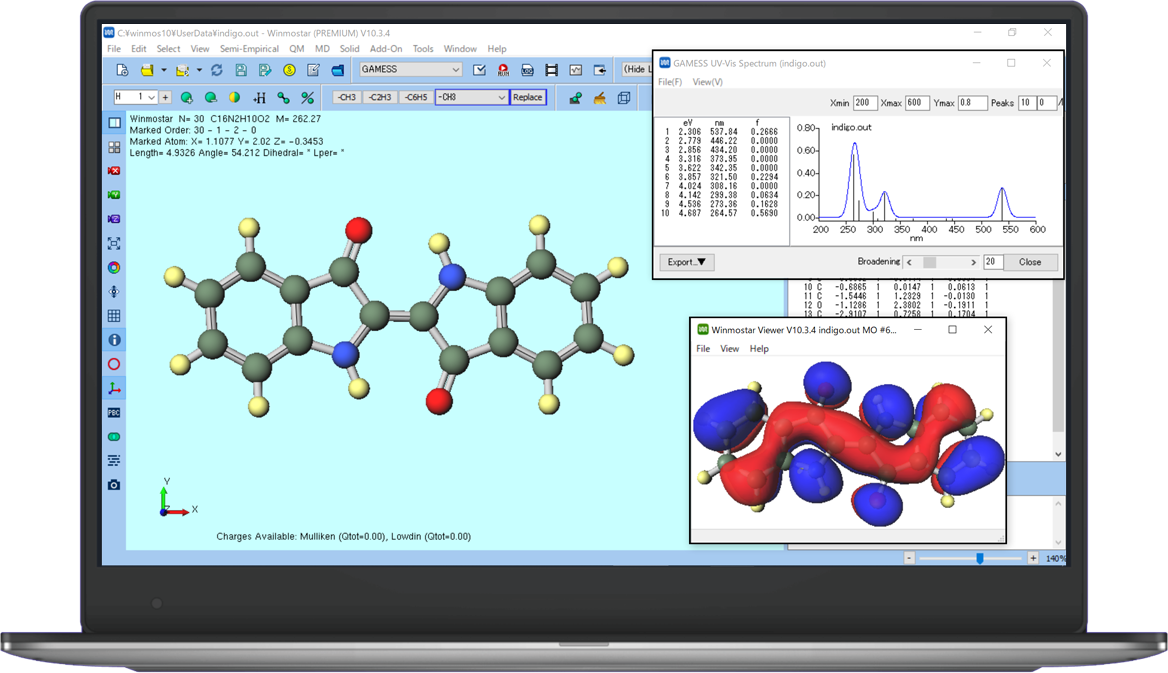
A unified GUI for pre- and post-processing GAMESS, NWChem, MOPAC, Quantum ESPRESSO, LAMMPS, and GROMACS — pro-tuned workflows that just work.
A unified GUI for pre- and post-processing GAMESS, NWChem, MOPAC, Quantum ESPRESSO, LAMMPS, and GROMACS — pro-tuned workflows that just work.
| All-in-One Workflow: Build structures, run simulations, analyze results, and visualize—all in one interface. |
| Works with globally adopted solvers: GAMESS, GROMACS, LAMMPS, MOPAC, Quantum ESPRESSO, and more. |
| Quick Solver Setup: Use our dedicated installer to get supported engines running in minutes—no complex configuration needed. |
| Run Where It Makes Sense: Control from Windows—run locally or submit to remote Linux/HPC solvers. |
| Comprehensive Learning Resources: Video tutorials, detailed manual, and FAQs guide you every step of the way. |
| Cost-Effective Performance: High quality at a lower price than legacy tools—flat licensing no matter how many cores you add. |
Deployed across manufacturing, education, and research; outcomes are documented in user-authored papers and patents. (As of Apr 30, 2026)
Countries
Universities Served
Citation in Academic Papers
Client Companies
Licenses Issued
Key Academic Users
Professional features for materials R&D, with audit-ready, reproducible workflows for patents and papers.
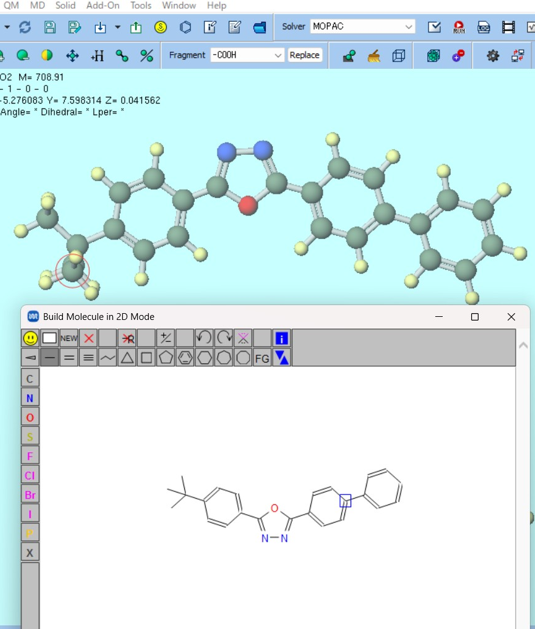
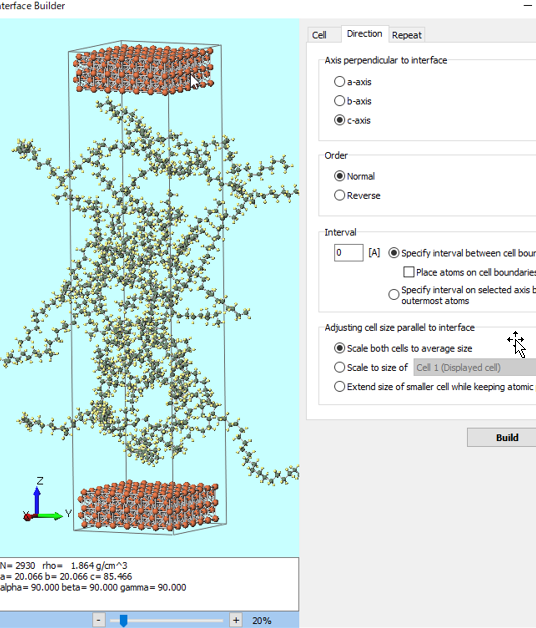
| Create diverse molecular and crystal structures. |
| Configure simulations with simple, flexible settings. |
| Seamlessly switch compute resources—local PCs or remote servers—as needed. |
| Automate file handling and process workflows. |
| Analyze results, visualize outputs, and compute multiple properties. |
| Convert and export simulation data in various formats. |
Extensive support ensures beginners feel confident.
FREE Edition
$0
STUDENT Edition
$0
Students only
PROFESSIONAL Edition Premium
$1,200〜
Educational Institution
$3,600〜
Industry/Government
PROFESSIONAL Edition Elite
$2,400〜
Educational Institution
$7,200〜
Industry/Government
