本ページのPDF版はこちら
量子化学計算、第一原理計算、分子動力学計算といった原子スケールのシミュレーション技術は、物質の研究開発における原理原則に基づいた手法の一つです。70年以上の歴史を持ちノーベル賞も受賞しており、学術界において正当な一手法として認められています(1998年、2013年)。主に
- 分析実験の補助
- 合成経路探索
- 現象のメカニズム解明
- 新規素材探索
などの場面で利用され、特許や論文での引用が増えており、産業界での利用も進んでいます。

図:"Density Functional Theory"と検索してヒットした各年の特許件数(Google Scholar)と論文数(Semantic Scholar)の推移
これらの技術は、エネルギーの計算方法(量子力学に基づくか、古典力学で近似するか)と物性の算出方法(運動から計算するか、静止構造から近似計算するか)で以下のように分類できます。
| 運動から物性を計算 (近似小・計算負荷大) |
静止構造から物性を計算 (近似大・計算負荷小) |
|
|---|---|---|
| 量子力学に基づきエネルギーを計算 (近似小・計算負荷大) |
第一原理分子動力学計算 | 量子化学計算・第一原理計算 |
| 古典力学に基づきエネルギーを計算 (近似大・計算負荷小) |
古典分子動力学計算 | 分子力学計算 |
なお、第一原理分子動力学計算は最も近似が少なく最も良さそうに見えますが、計算量が膨大なため極めて限られた原子数、時間スケールでしか実施できず適用できるケースが稀です。実際にはこれらの手法の中で、量子化学計算・第一原理計算、古典分子動力学計算が使われるケースが比較的多いです。
量子化学計算・第一原理計算
静止構造における電子状態を求め、そこから物性を算出します。量子化学計算と第一原理計算の違いが厳格に定義されているわけではありませんが、主に境界条件と基底関数の違いで分けられることが多く、前者は分子系に、後者は固体・表面系に適用されることが多いです(詳細はWinmostar基礎講習会で解説)。分子・結晶構造・電子状態・エネルギー・物性、各種スペクトル、化学反応における活性化状態などを取得します。(詳細はこちら)
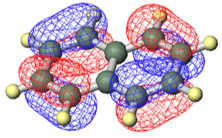
図:量子化学計算で得られたナフタレンのHOMO軌道の等値面
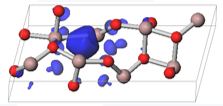
図:第一原理計算で得られたβ型酸化ガリウムの酸素欠陥準位の電荷密度の等値面
分子動力学計算
古典力学的に近似した原子間力を用いて原子の運動を求め、そこから物性を算出します。単に分子動力学計算と呼んだ場合には古典分子動力学計算のことを指す場合が多いです。分子の運動性、分子集合体の構造・熱力学量・物性などを取得します。(詳細はこちら)
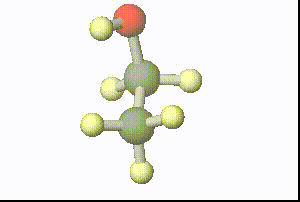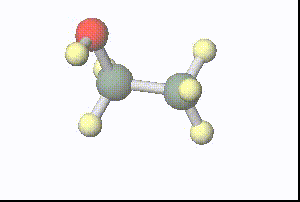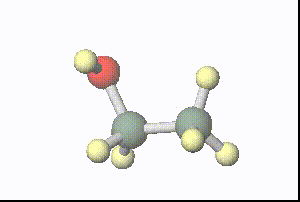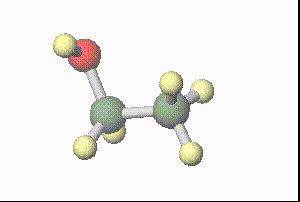
図:分子動力学計算で得られた1ピコ秒間の水溶媒中のエタノールの構造(溶媒は非表示)
運用における注意点とその解決方法の提案
これらの技術は、研究対象の現象のスケールと計算可能なスケールの差を接続する方法が物質や現象の種類ごとに大きく異なり、また現実的なコストで計算するために近似モデルが必要なため、容易に適用できない研究課題もあります。
Winmostarは様々な物質・現象を計算可能な機能を備えるだけでなく、経験豊富なスタッフが高品質なサポートを提供します(詳細・事例はこちら)。お客様の研究課題へ適用するためのコンサルティングや初心者でも安心の講習会・学習素材を提供します。そのため、これまでに341本の学術論文・19社の特許で利用実績があります。(2026年4月30日現在、詳細はこちら)
本HPでは各種のユーザ事例や100種類以上の手順書を公開しています。年6回無料開催しているWinmostar導入講習会にもご参加ください。
