6.16. menu
It is a menu related to Quantum ESPRESSO.
6.16.1. How to set up Quantum ESPERSSO
To install Quantum ESPRESSO, install CygwinWM version 2023/04/05 or later, which contains
pw.exe, etc. in/opt_win/QuantumESPRESSO*/bin/of CygwinWM. and so on in/opt_win/QuantumESPRESSO*/bin/.To use Quantum ESPRESSO from Winmostar, use Tools ‣ Preferences menu. If you use CygwinWM 2023/04/05 version or later, you do not need to configure.
First, under , select the Quantum ESPRESSO
pw.exethat you want to use. Next, under , select MPICH or Select and select thempiexec.exeof the MPI you want to use. If you are using Quantum ESPRESSO in CygwinWM, select/opt_win/MSMPI/Bin/mpiexec.exeunder CygwinWM. Then, under , enter the arguments for mpiexec.exe. If you use Quantum ESPRESSO in CygwinWM, enter -np % WM_NUM_PROC%. Finally, select the version ofpw.exethat you selected in , or 7.1 if you are using Quantum ESPRESSO in CygwinWM.If you want to add a pseudo-potential file, click Tools ‣ Preferences menu and add the pseudo Add the pseudo-potential file to the opened folder. The pseudopotential file extension must be upf (case insensitive). You can also create and select a new pseudo-potential folder.
If you want to add MOL files for RISM calculations, click Tools ‣ Preferences menu and add MOL files to the opened folder. The pseudopotential file extension must be mol (case-insensitive); you can also create a new folder for MOL files and select it.
Instructions for installing Quantum ESPRESSO on a remote machine can be found at Installing Winmostar and solvers.
6.16.2. Workflow Setup
Set up and run the Quantum ESPRESSO calculation flow in project mode.
- Preset
Import and saves a preset of settings.
- # of Jobs
Specify the number of jobs.
- Enable parameter/structure scan
This feature requires the purchase of an add-on. It allows you to run multiple calculations where only certain parameters differ (parameter scan) or to run calculations with the same parameters for multiple structures (structure scan).
Clicking :guilabel:Config opens the scan calculation settings screen.
For parameter scans, select %WM_SCAN1% for :guilabel:Target Variable, and enter the parameter values you want to assign to %WM_SCAN1% in each row of :guilabel:Values. Then, in the Workflow Settings window or the Keyword Settings window, enter %WM_SCAN1% for the parameter you wish to set. If multiple values are written in each row of Values separated by spaces, they can be referenced as %WM_SCAN1_1% for the first column, %WM_SCAN1_2% for the second column, and so on.
To scan the level of parallelism, select # of Threads or # of Procs.
For structure scans, ensure that an animation is displayed in the molecular display area (e.g., by opening an SDF file), and then select %WM_STRUCT% for :guilabel:Target Variable. In addition, when a structure scan is executed, the strings from the list in the animation display area are saved in the working folder as a file named label_scan_struct.txt.
After the scan calculation is finished, use to tabulate the calculation results.
- Import
Import the settings output by Export. Click the arrow at the right of the button to recall settings used in the past on the same project or Winmostar.
- Export
Output settings to file.
- OK
Runs a calculation or generates a file with your settings. See For project mode for details.
- Details
Set detailed calculation conditions. The Configure will be launched.
- Task
Select the type of calculation.
- Charge
Specifies the charge for the entire system.
- # of bands
Specify the number of bands.
- Spin
Set up the spin polarization calculation. Polarized (Manual) allows you to set the initial magnetization moment more flexibly for magnetic material calculations because Use Spin Density as is false and each element can have only one initial magnetization moment.
- Cutoff energy
Sets the cutoff energy (ecutwfc) of the wave function.
- Manually specify cutoff energy
If not checked, Cutoff energy is automatically specified from Precision.
- K points
Specify how to calculate K points.
- Pressure
Specify pressure.
- Phonon(DFPT)
Set up phonon calculations using DFPT(ph.x).
- Use Bravais-lattice index
If checked, input file will be created with other than ibrav=0. Check this box because it is necessary to set special points from ibrav when calculating band structure and phonon bands.
- Pseudopotential
Specify a pseudopotential file. changing Type will narrow down the Functional and Pseudo files. changing Functional will narrow down the Pseudo files. Changing Functional will narrow down the Pseudo files. See Pseudopotential in Configure for instructions if the desired choice does not appear.
- Properties
Calculate properties checked to match when calculating.
- Precision
Set calculation precision.
- Metal
Enable smearing. (occupations=smearing) Also adjusts the contents of Precision (above).
6.16.3. Configure
Set the calculation condition of Quantum ESPRESSO. To set up the calculations immediately after setting Run button, once to return to the main window please press :guilabel:` OK` button.
See Run for behavior when clicking Run.
Return to the default state with Reset button. Save the setting except Force Field with Save button. Load the setting saved by Save with the Load button.
If the Quantum ESPRESSO keyword displayed in the Keyword Editor and the contents set in this function window are different, you will be asked if you want to load the keyword from the Keyword Editor.
When invoking this function, if the structure displayed in the main window can be changed to a primitive cell, Convert Lattice (Primitive-Conventional) is executed automatically.
- Output Directory
Specify the output destination folder (outdir) of the data, and at the same time specify the new/continued execution of the job.
- Create
Create a new folder to output data to. outdir will be set to the new folder.
- Continue
Continue with the last executed QE job that is loaded on the main screen. outdir is set to the outdir of the last executed job.
- Select
Specify the folder specified in the dialog that opens as the output destination and continue the job from the data in that folder. outdir will be set to what you specify here.
- Preset
Select preset for setting. Each preset changes the following keywords
scf
scf
scf
relax
vc-relax
md
cp
scf
False
True
True
False
False
False
False
False
gamma
gamma
gamma
gamma
gamma
False
False
False
False
False
True
False
False
False
True
True
False
False
False
False
False
smearing
bfgs
bfgs
verlet
none
bfgs
False
False
False
True
True
True
True
True
False
False
False
False
True
False
False
True
False
False
False
False
True
False
False
False
second_order
second_order
sd
angstrom
angstrom
angstrom
angstrom
angstrom
crystal
False
True
True
False
False
False
False
False
False
True
True
False
False
False
False
False
False
True
False
False
False
False
False
False
False
False
True
False
False
False
False
False
- Use MPI
Specify whether to execute parallel calculation using MPI when executing QE. Enter the number of MPI processes in the horizontal field.
- Basic Tab
- restart_mode
When from_scratch is set, the job is started without using information from previous jobs; when restart is set, the job is started using information from outdir; when Automatically set, in project mode, from_scratch is used for new jobs and restart is used for other jobs. Automatically set is used in project mode for new jobs from_scratch, and restarts for all other jobs. In file mode, from_scracth is used when Output Directory=Create, and restart is used for other jobs.
- calculation
Select the type of calculation.
- # of bands
The selection in parentheses displays the number of valence electrons when pseudopotentials are used. If the valence number fails to calculate automatically, it is displayed as N/A.
- Do not specify
nbnd is set automatically.
- Specify nbnd
Explicitly specify the number of bands.
- Specify nbnd(Relative)
Specified relative to the number of valence bands.
- Use nbnd
Specify the number of bands. If not checked, it will be set automatically.
- K_POINTS
Choose how you want to specify the k-point from the pull-down menu and specify the k-point in the QE format in the text box below. If you want to use k-point information generated by a website such as SeeK-path, select the type of K_POINTS (e.g. crystal) and then paste the k-point coordinates and points into the text box below it. After selecting the type of K_POINTS (e.g. crystal), paste the coordinates of the k-points and the number of points into the text box below it. The number of k points (nks) written to the input file will be automatically filled in.
- gamma
Calculated at the
point only.
- automatic
Use Monkhorst-Pack method. Enter “(number of divisions in kx direction) (number of divisions in ky direction) (number of divisions in kz direction) (shift flag in kx direction) (shift flag in ky direction) (shift flag in kz direction)” (separated by spaces). The shift flag is 0 for no shift (k points contain
- automatic(by spacing)
Use Monkhorst-Pack method. The number of divisions in each direction is set by the Spacing parameter ( Spacing parameter ).
- automatic(by spacing,slab)
Use Monkhorst-Pack method. The number of divisions in each direction is set by the Spacing parameter ( Spacing parameter ). However, the number of vertical slab divisions detected automatically is set to 1.
- Others
Please refer to the pw.x manual or doc/brillouin_zones.pdf under the QE installation folder for details.
- Set default k-path
The default k-point path of the Bravo lattice (ibrav) detected for the crystal displayed in the main window is acquired from UserPref/kpath_default.txt and set.
- nosym
Specify whether to use spatial symmetry.
- noinv
Specify whether time reversal symmetry is used or not.
- Set ibrav = … and celldm
If the primitive cell is displayed in the main window and checked, set the appropriate ibrav and celldm. If it is not checked, set ibrav = 0 and set CELL_PARAMETERS.
- ecutwfc
Specify the cutoff energy of the plane wave when calculating the wave function.
- Ecut for US/PAW
Sets how ecutrho is specified when selecting an Ultrasoft or PAW pseudopotential file.
- Specify ecutrho/ecutwfc
Set ecutrho from the ratio of ecutrho to ecutwfc and the value of ecutwfc.
- Specify ecutrho
Directly set the value of ecutrho.
- ecutrho
Specify cutoff energy of plane wave at electron density and potential calculation.
- tot_charge
Specify the charge of the entire system in the simulation cell.
- occupations
Specify smearing for metals and tetrahedron for DOS calculations.
- ion_dynamics
Specify the algorithm to change ion (nucleus) position.
- cell_dynamics
Specify algorithm to change simulation cell.
- tprnfor
I will calculate the force.
- tstress
Calculate the pressure tensor.
- Advanced tab
- conv_thr
Specify tolerance of energy at SCF calculation.
- etot_conv_thr
Structural Optimization (relax) Specify the energy tolerance for calculation.
- forc_conv_thr
Structural Optimization (relax) Specify the force tolerance during calculation.
- press_conv_thr
Specify tolerance of pressure when cell structure optimization (relax - vc) calculation is calculated.
- electron_maxstep
Specify the maximum iteration number of the SCF calculation.
- nstep
Specify the maximum number of steps for structure optimization (relax) calculation and the number of steps for MD calculation.
- upscale
Specify coefficients for automatically decreasing conv_thr during structure optimization (relax) calculation.
- diagonalozation
Specify diagonalization algorithm.
- diago_david_ndim
Specifies the size of the workspace when diagonalized with the Davidson algorithm.
- spline_ps
Interpolate pseudopotentials with cubic spline, useful for GIPAW calculations.
- la2F (for pw.x)
Outputs a file with written eigenvalues for electron-phonon interaction calculations in pw.x.
- smearing
Specify the method of occupancy smoothing (smearing).
- degauss
Specify the parameter of occupancy smoothing.
- mixing_beta
Specify the mixture ratio of old and new KS orbitals in SCF calculation. The closer to 1, the greater the ratio of predicted values.
- mixing_mode
Specify the mixture algorithm of old and new KS orbitals.
- vdw_corr
Van der Waals (dispersion) Specify how to correct forces.
- Use input_dft
When checked, overwrite the setting of the functional to the setting written in the pseudopotential file.
- nqx1/2/3
Specifies the number of k-point sampling when calculating the Fock operator. If Default, use the same value as K_POINTS.
- cell_dofree
Specify the degree of freedom (direction) for moving the simulation cell.
- Use cell_factor
Explicitly specify the construction parameter of the pseudopotential table. Sometimes it is better to set a larger value for vc - relax (structure optimization calculation with cell size change).
- Spin/DFT+U tab
- nspin
Set up spin polarization calculation.
- Use tot_magnetization
When checked, specify the magnetization of the whole system here. If you do not check it, specify starting_magnetization.
- starting_magnetization
Gives the initial value of the magnetization of each atom species. Use “Use Spin Density as starting_magnetization” if you want to calculate antiferromagnets, since in this case all atoms of the same element have the same value.
- noncolin
Specifies whether or not non-colinear calculations are performed. If checked, nspin is not output to the input file. nspin is not output, but nspin=2 must be set on the same tab to set starting_magnetization.
- lspinorb
Pseudopotential file with spin orbit interaction can be used during calculation of non-colinear.
- Use Spin Density as starting_magnetization
Use the value of Spin Density set in the main window as-is as starting_magnetization. If checked, the above starting_magnetization is ignored. Use this feature when atoms of the same element have different starting_magnetization, such as in antiferromagnets.
- lda_plus_u
Perform LDA + U calculation.
- Hubbard_U/alpha
Specify the U and alpha parameters of Hubbard for each atom type.
- Phonon tab
- Run Phonon Calculation as Postprocess
Perform a phonon calculation. Specifically, ph.x is run after pw.x is run. To enable this item, you must select scf, nscf or relax for Calculation. If you want to run ph.x after the SCF or Relax calculation is finished, you can run it with Calculation=nscf, which will immediately finish pw.x and immediately start ph.x processing. The ph.x and other input/output files are created in the working folder.
- tr2_ph
Specify the censored error of phonon calculation.
- alpha_mix(1)
Specify mixing factor.
- epsil=.True.
Calculate the dielectric constant (electronic system) obtained from phonon calculation.
- lraman=.True.
Include calculation of Raman spectrum.
- electron_phonon
Specifies the method of electron-phonon interaction calculation.
- el_ph_sigma
Specifies the interval of double-delta smearing during electron-phonon interaction calculations.
- el_ph_nsigma
Specifies the number of double-delta smearing during electron-phonon interaction calculations.
- Use fildvscf
Output files used in electron-phonon interaction calculations when running q2r.x.
- la2F = .true. (for q2r)
Check in Electron Phonon Interaction Calculations
- asr(for dynmat)
Specifies how the Acoustic Sum Rule is given in the post-processing (dynmat.x) after phonon calculation. It does not affect the phonon calculation itself.
- lperm=.True.(for dynmat)
The contribution of the Gamma point to the dielectric constant (total dielectric constant including ionic vibrational contributions) is calculated in the post-processing (dynmat.x) after the phonon calculation.
- ldisp=.true.
Calculate phonon dispersion. In order to acquire phonon band structure, phonon density of states you need to specify this.
- nq1,nq2,nq3
Specify the number of K points when computing phonon variance.
- Use spacing
Use Monkhorst-Pack method. The number of divisions in each direction is set by the Spacing parameter ( Spacing parameter ).
- NMR/EFG tab
- Run GIPAW calculation (gipaw.x) as postprocess
Run a GIPAW calculation. Specifically, gipaw.x is run after pw.x is run. The gipaw.x input and output files will be created in your working folder. To see the details of the results of the GIPAW calculation, open gipaw.out or gipaw2.out directly in a text editor.
- job
Specify what you want the GIPAW calculation to output. If you choose ‘nmr & efg’, gipaw.x will be invoked twice, with job=nmr and job=efg, respectively.
- conv_threshold
Specify the truncation error for diagonalization.
- diagonalozation
Specify diagonalization algorithm.
- q_gipaw
Specifies the magnitude of the infinitesimal wavenumber to use when calculating with linear response theory.
- verbosity
Specifies the level of output to the log.
- use_nmr_macroscopic_shape/nmr_macroscopic_shape
Specifies whether the chemical shift values should be corrected to account for the macroscopic shape of the sample.
- spline_ps
Specifies whether pseudopotentials should be spline complement.
- q_efg
Specifies the quadrupole moment of the nucleus.
- hfi_nuclear_g_factor
Specifies the g-tensor of the nucleus.
- Enter q_efg
This is an input aid for q_efg. You can quote from the default or user-prepared q_efg tables.
- Open q_efg table
Open q_efg table.
- Dynamics tab
- Simulation Package
Specify the calculation package to be used for MD calculation. For cp.x, use the CPMD method.
- DT
Specify the time step of MD calculation with atomic unit.
- tempw
Specify the target temperature when temperature control is specified by MD calculation.
- press
Specify the target pressure when specifying pressure control in MD calculation.
- ion_temperature
Specify the temperature control method of ion (nucleus) in MD calculation.
- ion_velocities
Specify the initial velocity of ion for MD calculation.
- gangbang
Specify the allowable value of temperature during temperature control.
- pot_extrapolation
Born-Oppenheimer Specify the extrapolation method of the potential when using MD.
- wfc_extrapolation
Specify the extrapolation method of wave function when Born-Oppenheimer MD is used.
- electron_dynamics
Specify the algorithm to change the KS trajectory when Car - Parrinello MD is used.
- electron_velocities
Specify the initial speed of electrons when Car - Parrinello MD is used.
- emass
Car-Parrinello Specify the virtual mass of electrons when using MD.
- emass_cutoff
Car-Parrinello Specifies the cutoff value of the virtual mass of electrons during MD calculation.
- orthogonalization
Specify the method of matrix calculation (orthonormalization).
- Dipole Corr tab
- tefield
A sawtooth type external electric field is applied.
- dipfield
Use dipole correction.
- edit
Sets the direction in which the tefield and dipfield are applied.
- emaxpos
The location where the external electric field is at its maximum value when applying tefield and dipfield is given in fractional coordinates (in the range of 0 to 1).
- eopreg
The size of the region where the external electric field decays when applying tefield and dipfield is given in fractional coordinates.
- eamp
Gives the magnitude of the external electric field when applying the tefield and dipfield.
- ESM tab
- assume_isolated=esm
Check if you use ESM (Effective Screening Medium) method.
- esm_bc
Specify the type of boundary condition used in the ESM method.
- esm_efield
Specify the electric field.
- esm_w
Sets the offset for the location where the ESM will be placed.
- lfcpopt
Calculation of constant chemical potential (constant mu) will be carried out. The initial system charge is specified by tot_charge on the Basic tab.
- the fcp_m
Set the target value of Fermi energy in constant calculation of chemical potential.
- Enter Relative Potential
Supports input of Target Fermi Energy. First, specify the log file for calculation at voltage 0 and acquire Fermi energy at voltage 0. Next, input the applied voltage. From these two pieces of information, calculate Target Fermi Energy.
- RISM(1) tab
- trism=.True.
Enable RISM calculations. Check here to run ESM-RISM calculations, and check assume_isolated = esm on the ESM tab. To use this feature, you need to install a separate version of Quantum ESPRESSO with the ESM-RISM feature enabled.
- closure
Select the closure to use for RISM calculation
- tempw
Specify the initial velocity of ion for MD calculation.
- ecutsolv
Specify the cutoff energy of the plane wave when calculating the wave function.
- solute_lj
Specify the LJ parameter of the solute (DFT region). If none is selected, enter the LJ parameter in solute_epsilon and solute_sigma below
- noinv
Specify the number of molecular species of the solvent.
- SOLVENTS
Select the unit of Density from the pull-down menu and specify the density (concentration) of each solvent molecule type and the name of the MOL file. MOL files must be in the folder specified by Directory for MOL Files below.
- Directory for MOL Files
Specify a folder containing MOL files that can be selected with SOLVENTS.
- RISM(2) tab
- laue_expand_right/left
Specify the position of the far end of the solvent region in the ESM-RISM calculation.
- laue_starting_right/left
Specify the starting position of the solvent region in the ESM-RISM calculation.
- laue_buffer_right/left
Specify the location of the solvent buffer area in the ESM-RISM calculation.
- Run only 1D-RISM
If checked, run 1drism.x instead of pw.x. No DFT calculations will be performed. Useful if you only want to know the correlation function between solvent atoms and the chemical potential between solvents.
- rism3d_conv_level
Specify parameters for dynamically changing the truncation error of 3D-RISM calculation at each step of SCF calculation.
- rism1d/rism3d_maxstep
Specify the maximum number of 1D and 3D-RISM iterations.
- rism1d/rism3d_conv_thr
Specify the truncation error for 1D and 3D-RISM.
- mdiis1d/3d_size
Specify the convergence parameter of RISM calculation by MDIIS algorithm.
- mdiis1d/3d_step
Specify the convergence parameter of RISM calculation by MDIIS algorithm.
- Others tab
Fill in the other settings in QE's input file format (FORTRAN namelist format). An example of entry is displayed by pointing the pointer.
- Preview tab
A preview of the configuration keywords will be displayed.
- Options Tab
- Verbosity
Specify the amount of information output by QE.
- atomic_position unit
Specify the unit of ATOMIC_POSITIONS and CELL_PARAMETERS.
- Use max_seconds
If checked, processing of QE will be interrupted after the number of seconds entered here.
- Options for executable
Gives the arguments of the command to run pw.x or pw.exe. For example, you can specify -nk, -nb, -nt, or -nd to control parallelism.
- Dump all files for remote
Output files necessary for job execution under Linux environment. The same file as the file generated by the remote job submission function is output.
- Open k-path file
Open the configuration file (UserPref/kpath_default.txt) that describes the k-point path specified by default for each type of ibrav (bracket lattice). If UserPref/kpath_default.txt does not exist, it is copied from wmx/kpath_default.txt.
- QE Version
Specifies the version of the QE to which the input file of the output QE corresponds. The output format of some keywords (&fcp, HUBBARD, etc.) varies with the version.
- Properties tab
- Calculate these properties after pw.x
Select post processing to be executed immediately after executing pw.x. Various parameters of the processing selected here are specified in the Parameter/Value column on the right.
- Pseudopotential tab
- Mass
Specify the mass of each element.
- Default
Set standard mass.
- Light
Set the mass of all elements to 1. It is used for the purpose of promoting structural relaxation of ions.
- Manual
In the list below, for each element, the user individually sets the mass.
- Pseudopotential
Select a pseudopotential file from among those that are common to all elements in the system.
- (Type)
Select the type of pseudopotential.
- (Functional)
Selects the type of functionality. Choose from the choices narrowed down by (Type).
- (File)
Select a pseudopotential file. Choose from the choices narrowed down by (Type) and (Functional). If you choose (Manual), the user sets the pseudopotential individually for each element in the list below.
Pseudopotential files are searched in the folder specified in the pseudo Directory. Preference is given to the first entry in
qe_pseudo_priority_list.txtin the user configuration folder.- Reload pseudo Files
The pseudo-potential file placed in the folder specified by pseudo Directory is read again.
- pseudo Directory
Specify the folder where the pseudopotential file will be placed. If the case of pseudo in QE#39;s directory, use the pseudo folder under the installation directory of QE. If the case of (select …), use the directory selected in the dialog.
- Open Pseudo Directory
Open the folder specified in pseudo Directory.
- Download Pseudo Files
Download the pseudopotential file and install it.
- Open Priority List
Open UserPref/qe_pseudo_priority.txt. If it does not exist, it is copied from wmx/qe_pseudo_priority.txt.
- Reset
Reset settings.
- Import
Loading configuration file.
- Export
Output configuration file.
6.16.3.1. Spacing parameter
The Spacing parameter is used to determine the number of k-point divisions
in each direction, independent of the system size.
(where
is the index for each direction
) is the reciprocal lattice vector
for the lattice vector (basic translation vector)
.
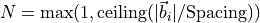
6.16.4. Run
Run Quantum ESPRESSO. The execution method differs depending on the situation.
When CPMD is selected cp.x, otherwise it executes pw.x.
(Default) When Output Directory = Create, create a new working folder and execute the calculation.
If the case of Output Directory = Continue, use outdir of the input file open in the main window as the working folder then.
If the case of Output Directory = Select, use the selected folder as the working folder (outdir).
Following file will be generated with execution. As an example, the file/folder name when the input file is
si.pwinis also shown.
type
Description
si.pwoutCalculation log file.
si.batIt is a batch file for running Quantum ESPRESSO.
si_qe_data\The following files are generated in the working folder. Only the main files are shown here.
type
Description
*.UPFpw_bands.inpw_bands.outpw_bands.in.pw_dos.inpw_dos.outpw_dos.inlog file.ph.inph.outph.in.Hint
** Working folder **
A working folder is a folder whose name is the name of the file opened in the main window plus a suffix.
** The suffix varies depending on the type of solver. **
For example, in the case of Gromacs, if the file opened in the main window is
aaa.groand the suffix is_gmx_tmp, the working folder will be namedaaa_gmx_tmp.It must be in the same hierarchy as the file opened in the main window.
Processing continues in the working folder of the same name even when continuing jobs, but by default the backup of the working directory of the previous job is created just before the continuation job is executed.
The name of the backup will be the one with the smallest number in the range where duplicate names do not exist. For example, if the working folder is
aaa_gmx_tmp, it isaaa_gmx_tmp1.** Directories without numbers are always up to date. **
Job is run through Winmostar Job Manager.
6.16.5. Open Log File (pwout)
Open the log file with a text editor.
6.16.6. Show Extracted Log
Excerpts of key information from the log file are displayed.
6.16.7. Animation
6.16.7.1. Optimization, BOMD(pwout, out)
From the information of the log file, animation such as structure optimization, BOMD calculation etc. is created and displayed.
For CPMD use CPMD(pos).
See Animation operation area for animation display operations. From the animation operation area, you can calculate radial distribution function, self-diffusion coefficient, mean square displacement, displacement of each atom, etc.
6.16.7.2. CPMD(pos)
Specify CPMD's pos and cel files and display animation.
To display the result of pw.x, use Optimization, BOMD(pwout, out).
See Animation operation area for animation display operations. From the animation operation area, you can calculate radial distribution function, self-diffusion coefficient, mean square displacement, displacement of each atom, etc.
6.16.8. Energy Plot
6.16.8.1. SCF Energy Change (pwout)
Select the log file and display a graph of total energy.
For CPMD use CPMD Energy Plot (evp).
- Property
Select the values to be displayed in the graph.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.8.2. CPMD Energy Plot (evp)
Specify the evp file of CPMD and display time variation of various energies.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9. Analyses
6.16.9.1. Density od States
Specify the working folder (outdir) and the SCF calculation log file and display the density of states.
Fermi energy is acquired from the log file of SCF calculation. dos.x runs in the background.
The range of energies where the density of states near the Fermi energy is zero is calculated internally, and the values of the band gap, VBM (upper end of the valence band) and CBM (lower end of the conduction band) energies are calculated and displayed.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.2. Projected DOS
Specify the working folder (outdir) and the SCF calculation log file and display projected density of state(PDOS).
Fermi energy is acquired from the log of SCF calculation. Projwfc.x runs in the background.
- Check atom using viewport
The molecular structure read from the log file is displayed in 3D. The orbitals of the atoms selected by clicking there are checked in the list of orbitals.
- Show band center
Calculate the band centers for each orbit. You can obtain the d-band center and others. You can choose whether to include the conduction band within the integration range.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.3. Band Structure
Specify the working folder (outdir) and SCF calculation log file and display the band structure.
Calculation must be completed with calculation = bands in advance. Fermi energy is acquired from the log file of SCF calculation. Bands.x runs in the background.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.4. Lowdin Charge
Specify the working folder (outdir), calculate and display the Lowdin charge.
Projwfc.x runs in the background.
6.16.9.5. Lowdin Charge
Specify a working folder (outdir) to calculate and display the Bader charge. You must generate a cube file of all electron densities beforehand using the PAW method.
The bader program is playing in the background.
6.16.9.6. Electron Density
Specify working folder (outdir) and display isoelectric density surface.
In the background, pp.x flows, and a cube file is generated.
Refer to Surface Setup / Cubgen window for how to operate subwindow.
6.16.9.7. Spin Density
Specify working folder (outdir) and display isoelectric density surface.
In the background, pp.x flows, and a cube file is generated.
Refer to Surface Setup / Cubgen window for how to operate subwindow.
6.16.9.8. Potential Energy
Specify a working folder (outdir) and display the isopotential energy surface.
In the background, pp.x flows, and a cube file is generated.
Refer to Surface Setup / Cubgen window for how to operate subwindow.
6.16.9.9. Potential Energy Distribution
Specify the working folder (outdir) and the log file of the SCF calculation to display the potential energy distribution in the z-axis direction (excluding exchange correlation energy).
Fermi energy is acquired from the log of SCF calculation. The difference between the Fermi energy and the maximum value of the potential energy ,excluding exchange-correlated energy, distribution is displayed as an estimate of the work function. Pp.x and average.x flow in the background.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.10. Fermi Surface
Specify the log file for SCF calculation and bands calculation and display the Fermi surface.
For Fermi surface display, use FermiSurfer <https://mitsuaki1987.github.io/fermisurfer/index_ja.html>. Specify the k point division number for bands calculation in # of K Points and press the Calc button to display the Fermi surface.
6.16.9.11. Dielectric Function
Specify the working folder after calculating the dielectric function and display the dielectric function.
- Direction
Specify the direction of the dielectric function to be acquired.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.12. NMR
Specify a working folder after GIPAW calculation to display NMR spectra.
See NMR Window for subwindow operation.
6.16.9.13. IR/Raman
Specify the working folder after phonon calculation and log of SCF calculation and display IR and Raman spectrum.
Refer to IR Spectrum Window for how to operate the subwindow.
6.16.9.14. Phonon Band Structure
Phonon Specifies the working folder after the variance calculation and displays the phonon dispersion curve.
- ASR
Specify the type of Acoustic Sum Rule to be applied.
- K Points
Specify the path of the dispersion curve to be acquired. In each line, describe the x, y, z components in units of 2pi/a, and next to it describe the number of divisions up to the next point. (Enter a total of 4 columns with a space separator)
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.15. Phonon Density of States
Phonon Specifies the working folder after distributed calculation and displays the phonon density of states.
- ASR
Specify the type of Acoustic Sum Rule to be applied.
- K Points
Phonon DOS Specifies the division number of K points during DOS calculation.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.16. Charge/Energy Profile (esm1)
Specify the esm1 file output by ESM calculation (assume_isolated = esm) and display the charge or energy distribution in the z-axis direction.
You can also plot the difference between the two esm1 files.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.17. 1D-RISM RMS Change
Plot the change in RMS of the 1D-RISM calculation performed at the beginning of the RISM calculation (trism = .True.).
- Draw
Displays a graph.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.18. 3D/Laue-RISM RMS Change for Last Step
Plot the change of RMS of 3D-RISM or ESM-RISM calculation in the last SCF step when RISM calculation (trism = .True.) is executed.
- Draw
Displays a graph.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.19. Solvent Pair Distribution Func. (1drism)
Interatomic correlation function (radial distribution function) of RISM region is calculated using 1 drism file outputted by RISM calculation (trism =. True.).
- Obtain Chemical Potentials
The chemical potential between solvent molecules is output in csv format.
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.9.20. Solvent Charge/Energy Profile (rism1)
Calculate the solvent density, energy, and charge in the direction in which the DFT region-RISM region contacts (vertical direction of the interface) by using the rism1 file output by RISM calculation (trism = .True.).
- Draw
Display the graph. The result analysis program is executed as necessary.
- Close
Close the window.
For how to operate the graph drawing area, see How to operate the graph.
6.16.10. Generate MOL File
Creates a MOL file for the solvent used in the RISM calculation (trism=.True.). To set LJ parameters from a LAMMPS data file, open the data file in the main window and check Use parameters in displayed file in the main window.
The created file should be placed in the folder specified by Directory for MOL Files on the RISM (1) tab of the Configure window of the MOL file.
6.16.11. Nudged Elastic Band
6.16.11.1. Workflow Setup/Keyword Setup
Set up and run the conditions for the NEB calculation; use
neb.xto run the NEB.Please set in the state that the structure optimization calculation of each of the start state and the end state has been completed.
6.16.12. Run
Run Quantum ESPRESSO NEB calculation in file mode.
6.16.12.1. Open Log File (pwout)
Open the NEB calculation log file in a text editor.
6.16.12.2. NEB error change
Displays errors in each iteration of the NEB calculation.
6.16.12.3. Animation
Specify the output file of the NEB calculation and display the changes in energy and atomic structure along the reaction coordinates after the NEB calculation convergence. If you want to see the energy change at each iteration of the NEB calculation continuously, use Energy Plot.
For the animation display operation method, see Animation operation area.
6.16.12.4. Energy Plot
Specify the output file of the NEB calculation and display the change in energy along the reaction coordinates. You can visualize the appearance at each iteration of the NEB calculation. Use Animation if you want to visualize the atomic structure.
6.16.13. BoltzTraP
BoltzTraP calculates thermoelectric characteristics based on the output of nscf calculation in QuantumESPRESSO.
6.16.13.1. Configure & Run
Set the calculation conditions for pre-post processing using BoltzTraP and execute pre-processing.
Create .intrans File button
Read nscf calculation output file (.pwout) of Quantum ESPRESSO and generate BoltzTraP setting file (.intrans). Assuming that the output file is mg2si_nscf.pwout, a working folder named mg2si_nscf is created in the same level. mg2si_nscf.intrans is generated in mg2si_nscf.
If the intrans file is successfully created, read the contents of the file and reflect it in the input fields of the following keywords.
- Fermilevel (Ry)
Fermi energy read from pwout file is set.
- energy grid
Specify the interval of the set energy.
- energy span
Specifies the range of band energies to consider in calculations around the Fermi level.
- number of electrons
Specify the number of electrons in a unit cell.
- lpfac
Specify the factor for complementing the band energy by Fourier expansion.
- efcut
Specify the calculation range by changing the chemical potential.
- Tmax
Specify the upper limit of the set temperature.
- temperature grid
Specify the interval of the set temperature.
- energy of bands
Specify the energy width of the band obtained from DOS.
- Calculate expansion coeff
If checked, compute expansion coefficients (CALC). If not checked, read expansion coefficients from file (NOCALC).
Start BoltzTraP button
Recreate the intrans file based on the setting conditions and execute BoltzTraP. At this time, the following files and folders are created.
The main files in the working folder (mg2si_nscf) at the end of the calculation are described below.
type
Description
mg2si_nscf.intransBoltzTraP input file.
mg2si_nscf.traceThis file contains information on energy and temperature dependence of thermoelectric properties calculated by BoltzTraP. The BoltzTraP Import Result menu reads this file and performs visualization.
Cancel button
Close Configure & Run window without doing anything.
6.16.13.2. Import Result
The following thermoelectric properties calculated by BoltzTraP are read and visualized.
Seebeck coefficient
Electrical conductivity
Electrical thermal conductivity
Power factor
Figure of merit
To display the energy dependence of the characteristic value at each temperature, select T [K] from the combo box and select the target temperature from the list. If you want to display the temperature dependence of the characteristic value for each energy, select E-Ef [eV] from the combo box and select the target energy from the list.
6.16.14. Phonopy
Calculations using Phonopy are performed in the following three steps.
Create a supercell based on a given crystal structure (project mode) or Quantum ESPRESSO input file (file mode).
Execute Quantum ESPRESSO for all generated super cells.
Create a ForceSets file from the Quantum ESPRESSO output file to calculate Phonon bands, DOS, thermophysical properties, etc.
6.16.14.1. Create a supercell
Generate a supercell for the Phonopy calculation for the crystal structure displayed in the molecule display area in the “Project” mode. Depending on the type of crystal, multiple structures may be generated. To run Quantum ESPRESSO calculations on these structures, use Workflow Setup with Enable scan calculation.
6.16.14.2. Configure & Run
In file mode, set the calculation conditions for pre- and post-processing using Phonopy and execute the pre-processing.
- Open button
Read the input file (*.in, *.pwin) of Quantum ESPRESSO. Post processing in Phonopy requires stress information. Therefore, the file to be read must include the tprnfor and tstress keywords. The input file of Quantum ESPRESSO for Phonopy can be set by using Quantum ESPRESSO Configure Preset=Phonopy.
- DIM
Specify the number of times the supercell repeats in the x, y, and z directions, separated by a space.
- MP
Specify the reciprocal lattice when calculating Phono DOS and thermodynamic properties in Phonopy, separated by a space.
- ATOM_NAME
Specify the elements included in the unit cell separated by a space. It is automatically entered when the input file is opened with the Open button.
Start button
Execute Phonopy based on the setting conditions, and create a super cell that is pre-processing. At this time, the following files and folders are created.
type
Description
si.batBatch file to execute the preprocessing of Phonopy.
si.shShell script file for executing the preprocessing of Phonopy.
si_ph_dataWorking folder.
The following files are generated in the working folder si_ph_data.
type
Description
mesh.confUsed to calculate density of states and thermodynamic properties in post processing of Phonopy.
band.confUsed when calculating band structure in Phonopy post processing.
header.insupercell-*.intmp-*.inCancel button
Close Configure & Run window without doing anything.
6.16.14.3. Edit .conf File
In file mode, opens the conf file in a text editor. Use this if you want to edit the keywords set in the keyword configuration screen.
6.16.14.4. Quantum ESPRESSO Continuous Execution
In File mode, Quantum ESPRESSO is executed on all super cells generated in the Keyword Setup and Execution screen. If you use this menu, Quantum ESPRESSO will run in the local environment.
6.16.14.5. Run Phonopy
Performs Phonopy post-processing in file mode.
At this time, the following files are created in the working folder :file: si_ph_data .
type
Description
phonopy.shShell script to execute Phonopy post processing
band.yamlInformation on the band structure calculated by post processing of Phonopy is output.
dos.datInformation on the density of states calculated by post processing of Phonopy is output.
thermal_properties.yamlInformation on thermodynamic properties calculated by Phonopy post processing is output.
6.16.14.6. Band Structure
Displays the band structure based on band.csv included in the working folder.
6.16.14.7. Density od States
Displays the density of states based on toal_dos.csv included in the working folder.
6.16.14.8. Thermodynamic properties
Displays thermodynamic properties based on thermal_properties.csv included in the working folder.
6.16.15. Elastic Constant Matrix
Calculate the elastic constant matrix. A structure scan calculation must be performed in advance on structures generated by setting Generate all strain modes in the Structure scan function.