6.13. menu
It is a menu related to LAMMPS.
6.13.1. How to set up LAMMPS
To install LAMMPS, install CygwinWM version 2023/04/05 or later, which contains
/opt_win/LAMMPS*/bin/inlmp_serial.exeandlmp _mpi.exeand so on.The configuration for using LAMMPS from Winmostar is done at Tools ‣ Preferences menu. If you have newly installed Winmostar V11.5.0 or later and are using CygwinWM 2023/04/05 version or later, no configuration is required.
First, select the LAMMPS
lmp_serial.exe,lmp_mpi.exeorlmp.exeto use with . (When you selectlmp_serial.exeto run MPI, thelmp_mpi.exein the same folder is automatically used.) Next, under , select MPICH or Select and choose the MPImpiexec.exeyou want to use. If you want to use LAMMPS stored in CygwinWM, select/opt_win/MSMPI/Bin/mpiexec.exeunder CygwinWM. Finally, at , enter the arguments for mpiexec.exe. If you use LAMMPS in CygwinWM, enter -np %WM_ NUM_PROC%.If you want to add a potential file, click Tools ‣ Preferences menu and in the opened folder Add potential files to the opened folder.
How to install LAMMPS on a remote machine is described in Installing Winmostar and solvers.
6.13.2. Assign Force Field
Set the force field. The choices vary depending on the type of solver.
After assigning a forcefield, use Get Info to check the assigned forcefield.
In the case of LAMMPS, if a gro file containing velocities is open in the main window at the time this function is used, a data file containing velocities is generated. Similarly, in the case of Gromacs, if a data file with velocities is open, a gro file with velocities is generated; this is useful when you want to take over Gromacs and LAMMPS calculation data with velocities.
Once you assign a force field and run the MD calculation, the bond order is automatically determined from the equilibrium length of the force field parameters. Depending on the type of force field, the bond order determined at that time may be different from the bond order before the force field assignment. Some force fields are affected by the bond order. Use Overwrite Bonds from File if you want to return to the bond order before force field assignment.
- Automatically assign parameters
Assign new force field parameters. Structures connected to each other by bonds in the molecule display area will be recognized as a single molecule.
- (General)
Specifies the force field for molecules other than proteins and water molecules. Internally, acpype is used for GAFF, GAFF2, OPLS-AA/L+GAFF, an in-house program for Dreiding, a proprietary extension of OpenBabel for UFF, and mktop for OPLS-AA. OPLS/AA-L+GAFF uses OPLS-AA/L for non-bonded potentials (such as Lennard-Jones parameters), while GAFF is used to define intramolecular bonded potentials (bonds, bond angles, dihedrals, etc.). The configuration for Dreiding is described in
polymer/dreiding.lib.txt. Check Universal Force Field for details on UFF.- Exception
For specific molecules or crystals, assign user-specified LJ parameters instead of using the force field selected in (General). Check the box for the molecule or crystal in the left column of the subwindow where you want to specify LJ parameters, then enter the LJ parameters in the right column. Clicking Automatically assign parameters automatically assigns UFF or Dreiding LJ parameters. Checking Use bond coefficient or Use angle coefficient sets harmonic bond and angle potentials for bonds or angles between atoms using the coefficient entered in the corresponding right column. These functions are intended only to approximate molecular or crystalline structures. :guilabel:If you do not use Use bond coefficient or Use angle coefficient, atoms constituting the specified molecule or crystal will interact solely via LJ or electrostatic potentials, potentially causing structural distortion.
Note
For example, when you want to allocate LJ parameters to solid phase atoms in a solid-liquid interface system.
- (Protein)
Specify the force field of the protein. Here, the atom to which the name of the amino acid residue is assigned in the PDB or gro format is recognized as a protein. Internally, gmx pdb2gmx is used.
Warning
This function can not be used when reading the molecular structure from a file not including residue name.
- (Water)
Specify the force field of the water molecule. You must specify the selected water model with Solvate/Build Cell. Internally we get the parameters from the library of Gromacs topology installed in Cygwin.
- Add [position_restraints] for protein
If a protein exists, write information (
[position_restraints]section) to constrain the position in the topology file with -POSRES on the Advanced tab. Ignored when protein is absent.- Add [position_restraints] for selected atoms
For the molecule specified by the user, write information (
[position_restraints]section) to constrain the position in the topology file with -POSRES on the Advanced tab. For example, when fixing solid phase in solid-liquid interface system.- Add [distance/angle/dihedral_restraints] for selected atoms
For the molecule specified by the user, write information to constrain distance, angle, dihedral angle to topology file by -POSRES on the Advanced tab.
- Open the Assignment Edit Window
After automatically assigning force field parameters, open the Edit Force Field window.
- Edit Force Field window
Enter force field parameters in Gromacs format within the table for atoms, bonds, angles, and dihedrals. To select an item from the molecular display area for use in the table, first click the desired atom (or the two atoms forming a bond, the three atoms forming an angle, and so on) within the molecular display area. Then click Select item clicked at viewport.
When this window is closed, 1–4 pairs are detected from the bond information, and [pairs] records for Gromacs are automatically generated. However, [pairs] are not generated for 1–4 pairs within four- or five-membered rings.
- File menu
- Import top file
Load the Gromacs top file and assign it to the displayed atoms.
- Export top file
Outputs the force field parameters currently being edited in Gromacs top format.
- Export mol2 file
Outputs the displayed molecular structure in mol2 format.
- Export itp file
Exports the force field parameters currently being edited in Gromacs itp format.
- Update using itp file (bond/angle/dihedraltypes)
Overwrites the force field parameters currently being edited with the bond types, angle types, and dihedral types in the specified itp file.
- Update using itp file (atom/bond/angle/dihedraltypes)
Overwrites the force field parameters currently being edited with the atom types, bond types, angle types, and dihedral types in the specified itp file.
- Action menu
- Complement
Assigns a specified type of generic force field to degrees of freedom that currently have no force field parameters assigned. Since multiple generic force fields may be assigned to a single molecule, please verify the calculation results in such cases.
- Assign equilibrium bonds/angles from current structure
Set the bond lengths and bond angles of the currently displayed molecule to the equilibrium lengths and angles of the force field parameters. The equilibrium lengths and angles assigned by the generic force field are carefully designed in conjunction with other parameters. Therefore, we recommend using this option only when the currently assigned equilibrium lengths and angles deviate significantly from the actual bond lengths and angles.
- Add undefined dihedrals
Add dihedral angles not defined in the dihedral table.
- Duplicate selected item
Duplicate the selected items in the table. This is useful when you want to create dihedral parameters with different periodicity.
- Delete selected item
Deletes the selected item in the table.
- Add unused dihedral terms
Use this when you want to create parameters with the specified periodicity for all dihedrals.
- Setup pair_style hybrid
This is only available for LAMMPS. Specify the force field defined by potential files such as EAM or Tersoff for the designated portion of the displayed molecules or crystal.
- Dump Now
Based on the current settings, generate a topology file.
Note
If you want to customize the forcefield information by editing it with a text editor, first save the file containing the forcefield information using Dump Now and edit the top for Gromacs or the data file for LAMMPS with a text editor.
Next, for Gromacs, import the gro file at (select Discard and import), then at Assign Force Field select :guilabel:` Select Use parameters written in topology file and click the OK button. You will then be asked for the location of the top file, so open the top file you just saved and edited.
For LAMMPS, import the data file at (select Discard and import), then at Assign Force Field, select Use the parameters written in file opened on :guilabel:`main window and click on the Next > button. If the force field information is not written in the data file, you will get a Choose the type of force field, choose the type of generic force field you want to use and click the OK button.
Charges are taken from the structure displayed in the main window. If more than one type of charge is set in the main window (for example, if the GAMESS log file is opened and Mulliken charge and Lowdin charge are set), the following order of priority is used: (high priority) User charge > NBO charge > Lowdin charge > ESP charge > Mulliken charge (low priority). When the file is opened and the Mulliken charge and Lowdin charge are set (for example, when the file is opened and the Mulliken charge and Lowdin charge are set), the order of priority is User charge > NBO charge > Lowdin charge > ESP charge > Mulliken charge (low priority).
- Use parameters defined in external parameter file (for inorganic system, ReaxFF or DPD)
(for LAMMPS) Select if you want to use the potential for inorganic materials, ReaxFF or DPD. After pressing the Next > button, specify the type of force field you actually want to use. pair_style and Potential file must be set in [Tools]-[Preferences]-[Calculations] to allow the user to enter them freely. A topology file is automatically generated for result analysis, but please note that it does not reflect the actual interatomic force parameters.
- Use parameters written in topology file
(For Gromacs) Select this option if you want to run MD calculations using a top file that already exists. The corresponding gro file must be opened or imported in the main window. If you edit the structure after opening or importing it, the correspondence with the top file will be broken and the calculation will not be possible. If you want to use this function after editing the structure after opening or importing it to the extent that it does not affect the force field information (for example, editing only the coordinates without changing the bonds), export the structure in gro format after editing it and open or import that file before using this function.
- Use parameters written in file opened on main window
(For LAMMPS) Select this option if you want to run the MD calculation using a data file that already exists. The main window must have the data file you want to use open or imported. If you edit the structure after opening or importing the file, the correspondence with the top file will be broken and the calculation will not be possible. After pressing the Next > button, specify the type of force field to use.
6.13.3. Workflow Setup
Set up and run the LAMMPS calculation flow in project mode. 12-Step Compression in Preset is the polymer equilibration procedure described in [Hofmann2000] , [Larsen2011] . Also, 21-Step Compression-Decompression is the polymer equilibration procedure described in [Larsen2011].
[Hofmann2000]
Hofmann, L. Fritz, J. Ulbrich, C. Schepers and M. Bohning, Macromol. Theory Simul., 9 (6), (2000), 293–327.
- Preset
Import and saves a preset of settings.
- # of Jobs
Specify the number of jobs.
- Enable parameter/structure scan
This feature requires the purchase of an add-on. It is possible to run multiple calculations in which only certain parameters differ (parameter scan) or to run calculations with the same parameters for multiple structures (structure scan).
Clicking Config opens the scan calculation settings screen.
For parameter scans, select %WM_SCAN1% for Target Variable, and enter the parameter values you want to assign to %WM_SCAN1% in each row of Values. Then, in the Workflow Settings window or the Keyword Settings window, enter %WM_SCAN1% for the parameter you wish to set. If multiple values are written in each row of Values separated by spaces, they can be referenced as %WM_SCAN1_1% for the first column, %WM_SCAN1_2% for the second column, and so on.
To scan the level of parallelism, select # of Threads or # of Procs.
For structure scans, ensure that an animation is displayed in the molecular display area (e.g., by opening an SDF file), and then select %WM_STRUCT% for Target Variable. In addition, when a structure scan is executed, the strings from the list in the animation display area are saved in the working folder as a file named label_scan_struct.txt.
After the scan calculation is finished, use to tabulate the calculation results.
- Import
Import the settings output by Export. Click the arrow at the right of the button to recall settings used in the past on the same project or Winmostar.
- Export
Output settings to file.
- OK
Run a calculation or generate a file with your settings. See For project mode for details.
- Details
Set detailed calculation conditions. The Configure will be launched.
- Ensemble
Specifies the type of ensemble.
- Temperature
Specify the temperature.
- Pressure
Specify the target pressure.
- Simulation time
Specify simulation time.
- # of snapshots
Specify number of dump and xtc outputs.
- Initial velocity
If Random, the first speed is generated randomly; if From parent, the last speed of the previous job is inherited.
- Free boundary condition
Calculate with free boundaries instead of periodic boundary conditions.
- Precision
Set calculation precision.
6.13.4. Configure
Set calculation condition of LAMMPS. To set up the calculations immediately after setting Run button, once to return to the main window please press OK button.
Behavior when clicking Run is see Run.
Assign Charges Automatically will be launched automatically if there is a molecule to which no charge is assigned. If no force field is assigned, Assign Force Field will be launched automatically.
Return to the default state with Reset button. Save the setting except Force Field with Save button. Load the setting saved by Save with the Load button.
- Continue Simulation
Execute a continuous job.
For details, see Run.
- Preset
Specify the preset of the calculation condition. Each preset changes the following keywords.
Pair style
lj/cut/coul/long
lj/cut/coul/long
lj/cut/coul/long
lj/cut/coul/long
Time step
2.0
2.0
2.0
# of time steps
5000
5000
5000
5000
Ensemble
minimize
nvt
npt
nve
True
False
False
Temperature
300
300
Pressure
1.0
Boundary Condition
p p p
p p p
p p p
p p p
Reset COM motion
linear
linear
linear
linear
Tchain
3
3
Pchain
3
Shake tolerance
1e-5
1e-5
1e-5
100
100
100
100
100
100
100
100
Log interval
10
10
10
10
Cutoff (vdW)
Cutoff (Coulomb)
PPPM order
4
4
4
4
K-space accuracy
1e-5
1e-5
1e-5
1e-5
Pair style
lj/cut/coul/long
lj/cut/coul/long
lj/cut/coul/long
lj/cut/coul/long
Time step
1.0
1.0
1.0
# of time steps
10000
10000
10000
10000
Ensemble
minimize
nvt
npt
nve
True
False
False
Temperature
300
300
Pressure
1.0
Boundary Condition
p p p
p p p
p p p
p p p
Reset COM motion
linear
linear
linear
linear
Tchain
3
3
Pchain
3
Shake tolerance
1e-6
1e-6
1e-6
200
200
200
200
200
200
200
200
Log interval
20
20
20
20
Cutoff (vdW)
Cutoff (Coulomb)
PPPM order
4
4
4
4
K-space accuracy
1e-6
1e-6
1e-6
1e-6
Pair style
lj/cut/coul/long
lj/cut/coul/long
lj/cut/coul/long
lj/cut/coul/long
Time step
0.5
0.5
0.5
# of time steps
20000
20000
20000
20000
Ensemble
minimize
nvt
npt
nve
True
False
False
Temperature
300
300
Pressure
1.0
Boundary Condition
p p p
p p p
p p p
p p p
Reset COM motion
linear
linear
linear
linear
Tchain
1
1
Pchain
1
Shake tolerance
1e-9
1e-9
1e-9
400
400
400
400
400
400
400
400
Log interval
40
40
40
40
Cutoff (vdW)
Cutoff (Coulomb)
PPPM order
K-space accuracy
Pair style
lj/cut/coul/cut
lj/cut/coul/cut
lj/cut/coul/cut
lj/cut/coul/cut
Time step
2.0
2.0
2.0
# of time steps
5000
5000
5000
5000
Ensemble
minimize
nvt
npt
nve
True
False
False
Temperature
300
300
Pressure
1.0
Boundary Condition
f f f
f f f
f f f
f f f
Reset COM motion
angular
angular
angular
angular
Tchain
3
3
Pchain
3
Shake tolerance
1e-5
1e-5
1e-5
100
100
100
100
100
100
100
100
Log interval
10
10
10
10
Cutoff (vdW)
Cutoff (Coulomb)
PPPM order
K-space accuracy
Pair style
lj/cut/coul/cut
lj/cut/coul/cut
lj/cut/coul/cut
lj/cut/coul/cut
Time step
0.5
0.5
0.5
# of time steps
20000
20000
20000
20000
Ensemble
minimize
nvt
npt
nve
True
False
False
Temperature
300
300
Pressure
1.0
Boundary Condition
f f f
f f f
f f f
f f f
Reset COM motion
angular
angular
angular
angular
Tchain
1
1
Pchain
1
Shake tolerance
1e-9
1e-9
1e-9
400
400
400
400
400
400
400
400
Log interval
40
40
40
40
Cutoff (vdW)
Cutoff (Coulomb)
PPPM order
6
6
6
6
K-space accuracy
1e-9
1e-9
1e-9
1e-9
Pair style
reax/c
reax/c
reax/c
reax/c
Time step
0.5
0.5
0.5
# of time steps
20000
20000
20000
20000
Ensemble
minimize
nvt
npt
nve
True
False
False
Temperature
300
300
Pressure
1.0
Boundary Condition
p p p
p p p
p p p
p p p
Reset COM motion
linear
linear
linear
linear
Tchain
1
1
Pchain
1
Shake tolerance
1e-9
1e-9
1e-9
400
400
400
400
400
400
400
400
Log interval
40
40
40
40
Cutoff (vdW)
Cutoff (Coulomb)
PPPM order
K-space accuracy
- MPI
Specify MPI parallel number.
- Basic
- Units
Specify the unit system.
- real
It is mainly specified by molecular system (A, fs, Kcal/mol).
- metal
It is mainly specified by crystal system (A, ps, eV).
- lj
It is mainly specified by DPD calculation (dimensionless unit).
- Atom Style
Specify the type of system to calculate. Units changes accordingly.
- Pair Style
Select the method of interaction calculation. lj/cut/coul/long and boundary is ppf, the command kspace_modify slab 3 is automatically added and the calculation is performed using the PPPM method considering the 2-D periodic boundary condition.
- Force Field/Potential File
If Units is real, specify the type of force field. Affects the special_bonds, bond_style, angle_style, dihedral_style, and improper_style keywords.
Select the potential file when Units is other than “real”. List the files in the
Potentialfolder directly under the folder where you installed the LAMMPS main unit. The choices will change according to Pair Style.- Time Step
Specify the step size of time integration. Units are selected according to the selected Unit.
- # of Time Steps
Specify the maximum number of time integration steps.
- Ensemble
Specifies the type of time integration. Select one of the following:
nvt(canonical ensemble with constant temperature),npt(ensemble with constant temperature and pressure),nve(microcanonical ensemble with constant volume and energy),minimize(energy minimization using the conjugate gradient method), ornve/limit(similar to nve, but with a limit on the displacement per step). nve/limit option is used as an alternative to structural optimization (minimize).- Generate Velocity
If you check, the initial speed will be given.
- Random Seed
Specify the seed of the pseudorandom number at the time of initial velocity occurrence.
- Temperature
Specify the target temperature. At the time of annealing calculation, specify the temperature of the start condition.
- Tdamp
Specify the time constant parameter for temperature control.
- Use berendsen thermostat
Use fix temp/berendsen instead of fix nvt or npt to control temperature.
- Use velocity rescaling
Use fix temp/rescaling instead of fix nvt or npt for temperature control.
- Pressure Control
Specify how cells are moved during pressure control.
- Pressure
Specify the target pressure.
- Pdamp
Specify the time constant parameter of pressure control.
- Use berendsen barostat
Use fix press/berendsen instead of fix nph or npt to control pressure.
- Advanced
- Boundary X Y Z
Specify the periodic boundary condition.
p(periodic),f(non-periodic and fixed),s(non-periodic and shrink-wrapped),m(non-periodic and shrink-wrapped with a minimum value). If pair_style is lj/cut/coul/long and boundary is ppf, the command kspace_modify slab 3 is automatically added and the calculation is performed using the PPPM method considering the 2-D periodic boundary condition.- Energy Tolerance
minimize Specifies the truncation error on energy during calculation.
- Force Tolerance
minimize Specifies the truncation error on force during calculation.
- Reset COM Motion
In the case of xy, only the center-of-gravity motion is frozen in the x and y directions, but not in the z direction.
- Reset Interval
Specify Reset COM Motion frequency in time step
- Max distance for nve/limit
Specifies the maximum displacement per step for Ensemble = nve/limit.
- Tchain
Specify the number of stages of Nose-Hoover chain.
- Pchain
Specify the number of stages of pressure control.
- Velocity rescaling interval
Specify how often to apply speed scaling.
- Velocity rescaling window
Specify the window (allowable range from set temperature) when applying speed scaling.
- Velocity rescaling fraction
Specifies the fraction at which to apply speed scaling; if 1.0, it will match the set temperature immediately after application.
- Control temperature of specific group only
This function controls only the temperature of a specific group. To do this, register a group to be controlled with Select Registered Group and press Select Group button to select the group.
- Constrain hydrogen atoms
We restrict hydrogen atoms by SHAKE method.
- SHAKE tolerance
Specify the truncation error of the SHAKE method.
- Automatically disable Shake if CH4-like molecule exists
Automatically disables the SHAKE method when methane like molecules are included.
- Set “box tilt large”
Specify the allowable degree of deformation of the simulation cell.
- Enable dynamic load balancing
Enable dynamic load balancing.
- Output
- Dump Interval (dump)
Specify the frequency of outputting coordinates in dump format as the number of time steps.
- Dump Interval (xtc)
Specify the frequency of outputting coordinates in xtc format as the number of time steps.
- Dump Interval (xyz)
Specify the frequency of outputting coordinates in xyz format by time step number.
- Dump Interval (restart)
Specify how often to output the restart file, in number of time steps.
- Log Interval
Specify the frequency of writing energy variables to the log file by time step number.
- Print log in high precision
Increases the number of digits of the energy variable to be written to the log file.
- Sort dump file by id
Makes the order of particles in the dump file sorted by id (consecutive number).
- Flush log
Flush each time when the log is output.
- Include velocities in dump custom
When outputting in dump format, also output the velocity.
- Include forces in dump custom (minimize only)
Only when Ensemble=minimize, forces are also output when writing in dump format.
- Extra variables for log
Add any variables to thermo_style.
- Dump interval (dipole)
Specify the frequency of outputting time series data of dipole moments for the entire system in terms of the number of time steps.
- Calculate Fluctuation Properties
It calculates and outputs the heat capacity and isothermal compressibility on the fly from fluctuations in thermodynamic quantities. In the Energy Plot feature, selecting v_Cvm displays the cumulative average value of the constant-volume heat capacity, selecting v_Cpm displays the cumulative average value of the constant-pressure heat capacity, and selecting v_betat displays the cumulative average value of the isothermal compressibility.
- Calculate Thermal Conductivity
Calculate and output the thermal conductivity on-the-fly from the autocorrelation function of the atomic flow velocity and the Green-Kubo equation. This method uses the fix ave/correlate command, so the length of the autocorrelation function is fixed. In the Energy Plot feature, selecting v_kappa displays the thermal conductivity obtained up to that point in time.
- Calc interval
Specifies how often the autocorrelation function is calculated.
- ACF length
Specifies the length of the autocorrelation function. The maximum time for an autocorrelation function is (Calc Interval)×(ACF Length)×(Time Step).
- Calculate viscosity
Calculates and outputs viscosity on-the-fly from the autocorrelation function of the pressure tensor and the Green-Kubo equation. This method uses the fix ave/correlate command, so the length of the autocorrelation function is fixed. In the Energy Plot feature, selecting v_eta displays the viscosity obtained up to that point in time.
- Calc interval
Specifies how often the autocorrelation function is calculated.
- ACF length
Specifies the length of the autocorrelation function. The maximum time for an autocorrelation function is (Calc Interval)×(ACF Length)×(Time Step).
- Calculate heat flux relaxation
Output autocorrelation functions for atomic heat flow. This method uses the fix ave/correlate/long command (multiple-tau correlator), so you can automatically get long-time autocorrelation functions depending on the simulation time. Thermal conductivity can be calculated separately from post-processing.
- Calc interval
Specifies how often the autocorrelation function is calculated.
- Dump Interval
Specifies the output frequency of the autocorrelation function.
- Calculate stress relaxation
Output autocorrelation function for the pressure tensor. This method uses the fix ave/correlate/long command (multiple-tau correlator) so you can automatically get long autocorrelation functions depending on simulation time. Viscosity can be calculated separately from post-processing.
- Calc interval
Specifies how often the autocorrelation function is calculated.
- Dump Interval
Specifies the output frequency of the autocorrelation function.
- Calculate dipole relaxation
Outputs the autocorrelation function of the dipole moments for the entire system. This method uses the fix ave/correlate/long command (multiple-tau correlator) so you can automatically get long-time autocorrelation functions depending on simulation time.
- Calc interval
Specifies how often the autocorrelation function is calculated.
- Dump Interval
Specifies the output frequency of the autocorrelation function.
- Output (2)
- Dump velocity profile along
Outputs the average velocity distribution along the specified axis. The output can be viewed in Axial velocity distribution.
- Calc interval
Specify the output frequency of the average velocity distribution.
- Dump interval
Specifies the output frequency of the autocorrelation function.
- Slab width
Specify the thickness to be spatially averaged when calculating the average velocity distribution.
- Target
Select the direction of the velocity to sample when calculating the average velocity distribution.
- Interaction
- Modify cutoff radii not to exceed L/2
If checked, Cutoff(vdw) and Cutoff(Coulomb) are automatically adjusted so that they do not exceed half the lattice constant.
- Neighbor Search
Specify algorithm for near particle search.
- Neighbor Skin
Specify the extra radius of the search radius when searching for nearby particles.
- Cutoff(vdw)
vdw (LJ) Specifies the cutoff radius of the potential.
- Enable Long Range Correction
Specify the presence or absence of the vdw potential cut-off correction term.
- Cutoff(Coulomb)
Coulomb (electrostatic) Specify the cutoff radius of the potential.
- Disable Ewald(PPPM) if no charge exists
Automatically disables the Ewald (PPPM) method when the system has no charge.
- Automatically set Nmesh
The number of meshes of the PPPM method used when Pair Style =
lj/cut/coul/longis automatically set from K-space accuracy.- Nmesh for kx, ky, kz
Specify the mesh number of PPPM method.
- PPPM Order
Specify Spline interpolation order of PPPM method.
- K-space accuracy
Specify the allowable relative error of PPPM method.
- Non-equiliibrium (1)
- Enable Elongation
Enable decompression calculation. Ensemble can be specified when it is not
minimize. In the Energy Plot feature, selecting v_EngStrain allows the engineering strain to be displayed.- Affine Transformation
Specify whether to modify the atom position according to the simulation cell and to affine (similarity) deformation during elongation calculation.
- Eng. Strain Rate
Specify the extension speed at extension calculation with industrial strain. Max Eng. Strain shows the predicted value of the strain at the final step.
- Preserve Volume
During elongation calculation, deform the cell size in the direction perpendicular to the elongation direction so that the volume of the simulation cell is kept constant.
- Enable Pulling
Enable Pull calculation to move a specified atom group at a constant speed. Ensemble can be specified when it is not
minimize.- Pulled Atoms
Press Select Group button and select the group in the state where the atoms you want to pull are registered by Select Registered Group in advance.
- Pull Velocity
Specify the pull speed for Pull calculation.
- Enable Simulated Annealing
Enable annealing calculations (calculation to change the temperature at a constant speed). Ensemble can be specified when
nvt,npt. The value of Temperature is the temperature at the beginning, and the value of Final Temperature is the temperature of the final state.- Final Temperature
Specify the temperature of the final state at the time of annealing calculation.
- Annealing Rate
The heating or cooling rate at the time of annealing calculation is displayed.
- Enable pressurization
Enable annealing calculations (calculation to change the temperature at a constant speed). Ensemble can be specified when
nvt,npt. The value of Temperature is the temperature at the beginning, and the value of Final Temperature is the temperature of the final state.- Final Pressure
Specify the temperature of the final state at the time of annealing calculation.
- Enable electric field
Apply an external electric field. Selecting Sine wave applies the electric field using a sine function. Selecting Cosine wave applies the electric field using a cosine function.. Constant applies a steady electric field. In Energy Plot, the electric fields applied by this feature are displayed as v_efieldx, v_efieldy, v_efieldz, and the corresponding response, the overall dipole moment of the system, is displayed as v_dipolex, v_dipoley, v_dipolez.
- Amp & Freq
Specify the intensity (Amp) and frequency (Freq) for each direction. When Enable electric field is selected and Sine wave or Cosine wave is chosen, the electric field is given by the following equation, where A is the intensity and f is the frequency (unit: 1/fs = PHz, petaHertz). When Constant is selected, only the intensity is used.
- Non-equiliibrium (2)
- Enable adding force
Target atoms applies a force in the z-direction to the atoms specified in Target atoms. The force applied is the value of Target Pzz and the area of the XY plane of the simulation cell. In solid-liquid interfacial systems, this is useful when you want to apply a force to a solid (slab) and adjust the pressure throughout the system. The position of the center of gravity in the z-direction of the atom applying the force is output to thermo_style in the variable v_GrpAddForceCOMz and can be monitored using Energy Plot.If control is not working properly, setting Target Pzz to a high value (e.g., 10000 atm) and then gradually decreasing it may resolve the issue. Additionally, specifying structures like single-layer graphene as Target atoms can cause the calculation to fail. Therefore, it is recommended to specify only a portion of the atoms, such as the top or bottom edge of the slab, as Target atoms.
- Target atoms
Press the Select Group button with the atoms you want to force registered beforehand with Select Registered Group and select that group.
- Target Pzz
Enable adding force
- Enable direct density control
Force the simulation cell to be linearly deformed so that the density is at the value of Density at final step at the final step. The deformation is analogous. This is useful when the pressure control is not stable but you want to compress the system to some extent.
- Density at final step
:guilabel:Specifies the density of the final step in the Enable direct density control feature.
- Enable SLLOD method
This enables SLLOD method calculations. In the Energy Plot feature, selecting v_etane displays the instantaneous value of the viscosity, and selecting v_tempSLLOD displays the instantaneous temperature value with the SLLOD bias removed.
- Shear strain rate
Specifies the shear rate for SLLOD method calculations.
- Divide # of time steps by shear strain rate
Set the number of time integration steps to the value obtained by dividing the shear strain rate. When running calculations with varying shear strain rates, using this feature allows you to reduce the number of steps for high shear strain rates and increase the number of steps for low shear strain rates without explicitly changing the # of time steps.
- Pulling with thermostat
This function controls the specified atomic group toward an average position or velocity while allowing thermal motion via a heat bath. In the Restraint and Pulling functions, the temperature of the controlled group becomes 0 K, whereas this function allows control at a specified temperature. In the Energy Plot feature, selecting v_PullTherm*_rx, v_PullTherm*_ry, v_PullTherm*_rz displays the x, y, z coordinates of the center of mass of the controlled group; selecting v_PullTherm*_vx, v_PullTherm*_vy, v_PullTherm*_vz displays the x, y, z components of the center-of-mass velocity; selecting v_PullTherm*_fx, v_PullTherm*_fy, v_PullTherm*_fz displays the x, y, z components of the force acting on the center of mass; and selecting v_TempPullTherm*COM displays the instantaneous temperature of the controlled group.
- Restraint
- Enable Restraint
Calculation is performed by constraining the distance between specified two atoms. Ensemble can be specified when it is not
minimize.- Restrained Atoms
When you click the Set button, the two atoms with the markers become the target of the constraint.
- Bond Length
Specify the constraint distance between two atoms at the time of constraint calculation.
- Initial Strength
Specify the spring coefficient of the constraint potential in the starting state at the time of constraint calculation.
- Final Strength
Specify the spring coefficient of the constraint potential in the final state at the time of constraint calculation.
- Enable Position Restraint
Calculate with the absolute coordinates of the specified atom fixed. The temperature of the unfixed atom is output to the log as TempFree.
- Restrained Atoms
Press Select Group button to select the group with the atoms to be constrained registered with Select Registered Group in advance, .
- Use spring potential
If Restrained Atoms is checked, the atoms will be restrained by the spring potential from their initial position. If Reset positions of restrained atoms after run is checked, the atoms will be returned to their initial positions after the calculation is finished, so they will not move away from their initial positions even if you repeat the continue simulation. If the Restrained positions of atoms after run checkbox is checked, the position will be returned to the initial position after the calculation.
- Spring constant
Specify the spring coefficient when using the spring potential.
- Reset positions of restrained atoms after run
When using the spring potential, the position of the constrained atoms is reset to the initial position after the calculation is complete.
- Automatic
- Rescale velocities to..
Use it when you want to bring the system temperature closer to the target temperature in the NVE ensemble. Calculate the scaling factor from the average temperature under calculation and the temperature entered here and scale the velocity of each particle in the final structure.
- Rescale cell size to..
It is used when calculating with the NVE or NVT ensemble in the state close to the set pressure after calculating with the NPT ensemble. Scale the final structure to the average cell size under calculation.
- Additional Commands
Add any command just before the read_data line, after the various fix commands, just before the run (or minimize) line, and just after the run (or minimize) line.
- Manual entry
The contents of the generated LAMMPS input script (in file) will be displayed. You can also edit directly at this location. Any additional information you add here will be discarded when you edit other keywords. If you want to avoid this, fill in the Additional Commands field.
- Options
- Restore Working Folder
Click this button to return the working folder to the state before execution, such as when the continued job terminates abnormally.
- Dump all files for remote
Output files necessary for job execution under Linux environment. The same file as the file generated by Remote job function is output.
- Generate gro & ndx files every time
If it is not checked, gro and ndx files will not be generated for continuous jobs.
- Extra File
Click Add to copy the selected files to the working folder; click Clear to clear the added files.
- Reset
Reset settings.
- Import
Loading configuration file.
- Export
Output configuration file.
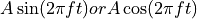
6.13.5. Run
Execute LAMMPS. The execution method differs depending on the situation.
- (Default) Continue Simulation is unchecked and Automatically assign parameters or Use parameters defined in external parameter file (for inorganic system, ReaxFF or DPD) in Assign Force Field is selected
Create a new data file (file containing coordinates and topology) and start the job.
- Continue Simulation is unchecked and Use parameters written in file opened on main window” in Assign Force Field is selected
Start the job using the data file opened in the main window.
- When Continue Simulation is checked
Start the job using the
lmp_tmp_final.datalocated in the working folder associated with the data file opened in the main window.Following file will be generated with execution. As an example, the file/folder name when the input file is
water.datais also shown.
type
Description
water.logThis is the log file of LAMMPS.
water.batwater_lmp_tmp\The following files are generated in the working folder. Only the main files are shown here.
type
Description
lmp.datalmp.inlmp.loglmp.dumplmp.restartlmp_final.datapostproc.shlmp_tmp_final.datagenerated by LAMMPS is not sufficient to run LAMMPS as is, so this script does some processing to make up for the insufficient information.lmp.xtclmp.grolmp.toprestart.batrestart.inHint
**Working folder**
A working folder is a folder whose name is the name of the file opened in the main window plus a suffix.
The suffix varies depending on the type of solver.
For example, in the case of Gromacs, if the file opened in the main window is: file: aaa.gro and the suffix is
_ gmx_tmp, the working folder will be namedaaa_gmx_tmp.It must be in the same hierarchy as the file opened in the main window.
Processing continues in the working folder of the same name even when continuing jobs, but by default the backup of the working directory of the previous job is created just before the continuation job is executed.
The name of the backup will be the one with the smallest number in the range where duplicate names do not exist. For example, if the working folder is
aaa_gmx_tmp, it isaaa_gmx_tmp1.Directories without numbers are always up to date.
The job is run through Winmostar Job Manager.
6.13.6. Open Log File
Open the LAMMPS log file (
*.log) with a text editor.
6.13.7. Show log excerpts
Displays excerpts of key information from the log file.
6.13.8. Animation
Select the data file and dump file and animate the MD calculation trajectory.
The file name of the main window does not change.
For the animation display operation method, see Animation operation area.
In the case of calculations where changes in chemical bonds occur, such as ReaxFF, checking the checkbox in the Animation Manipulation Area will determine whether or not a bond exists based on the bond distance at each step, allowing you to see how the bond changes. You can check how the bond changes.
Click Open Viewer in the animation operation area, and then in Winmostar Viewer, change Color to Property from . For energy minimization (minimize) calculations, this displays the common logarithm of the absolute value of the force acting on each atom in color. For other calculations, it displays the absolute value of each atom’s velocity in color.
6.13.9. Energy Plot
Select the log file and display a graph of various thermodynamic quantities such as energy, temperature and pressure. You can plot the value specified by thermo_style. v_logFn is the common logarithm of the maximum force in energy minimization (minimize) calculations.
Please see Energy Plot window for how to operate subwindow.
6.13.10. Import Last Coordinate (data)
Open
*_lmp_tmp\lmp_tmp_final.gro.When using this function, the file name of the main window does not change.
6.13.11. Analyses
6.13.11.1. Radial Distribution Function
Select the xtc file output by LAMMPS and the gro, ndx file automatically generated by Winmostar and display the radial distribution function.
See Radial Distribution Function for details.
6.13.11.2. Diffusion Constant/Mean Square Displacement
Select the xtc file output by LAMMPS and the gro, ndx file automatically generated by Winmostar, and display the mean square displacement and the self diffusion coefficient.
See Diffusion Constant/Mean Square Displacement for details.
6.13.11.3. Scattering Function
Select the xtc file output by LAMMPS and the gro, ndx file automatically generated by Winmostar and display the scattering function.
For details, see Scattering Function.
6.13.11.4. Total Linear Moment
Select the xtc file output by LAMMPS and the gro, ndx file automatically generated by Winmostar and display the total linear moment.
See Total Linear Momentum for details.
6.13.11.5. Velocity Distribution
Select the xtc and dump files output by LAMMPS, along with the gro, ndx, mdp, top, and data files automatically generated by Winmostar, and display the velocity distribution.
See Velocity Distribution for details.
6.13.11.6. Velocity Correlation / Vibration Spectrum
Select the xtc file output by LAMMPS and the gro, ndx, mdp, and top files automatically generated by Winmostar, and display the velocity autocorrelation function and vibrational spectrum.
See Velocity Autocorr/Vibration Spectrum for details.
6.13.11.7. Static Dielectric Moment
Select an xtc file output by LAMMPS and a gro, ndx, mdp, or top file automatically generated by Winmostar to display the distribution and histogram of relative permittivity or dipole moment.
See Radial Distribution Function for details.
6.13.11.8. Density Profile
Select the xtc file output by LAMMPS and the gro, ndx, mdp, and top files automatically generated by Winmostar, and display the density distribution.
See Radial Distribution Function for details.
6.13.11.9. Free Volume
Select the xtc file output by LAMMPS and the gro, ndx, mdp, and top files automatically generated by Winmostar, and display the free volume.
See Free Volume for details.
6.13.11.10. Axial velocity distribution
Select the information file containing the axial average velocity distribution output by LAMMPS to display the axial velocity distribution.
6.13.11.11. Various autocorrelation functions (ave/correlate)
Displays the autocorrelation function created by the fix ave/correlate command, which is output when calculating thermal conductivity and viscosity using the Green-Kubo formula
6.13.11.12. Various autocorrelation functions (ave/correlate/long)
Displays the autocorrelation function created by the fix ave/correlate/long command.
6.13.11.13. Hildebrand solubility parameter
The Hildebrand solubility parameter is calculated from the log and data files output by LAMMPS. Calculation results for both the gas phase and the liquid phase are required. To obtain the compressibility needed for calculating DPD parameters, the LAMMPS calculation that generated the files to be read must have Calculate Fluctuation Properties enabled.
6.13.11.14. Chi/DPD parameters
The Chi parameter and the DPD aij parameter are calculated from the log and data files output by LAMMPS. Calculation results for both the gas phase and the liquid phase of the two components are required. Internally, the values calculated in Hildebrand solubility parameter are used.
6.13.11.15. Bond/Angle/Dihedral Distribution
Select the xtc file output by LAMMPS and the gro, ndx, mdp, and top files automatically generated by Winmostar, and display the distribution of distances, angles, or dihedral angles between the selected groups.
For details, see Scattering Function.
6.13.11.16. Hydrogen bonding analyses
Select the xtc file output by LAMMPS and the gro, ndx, mdp, top files automatically generated by Winmostar, and analyze hydrogen bonds between the selected groups.
See Radial Distribution Function for details.
6.13.11.17. radius of inertia
Select the xtc file output by LAMMPS and the gro, ndx, mdp, top files automatically generated by Winmostar, and analyze the radius of gyration of the selected group.
See Radius of Gyration for details.
6.13.11.18. Atom/Group Distance Change
Select the xtc file output by LAMMPS and the gro, ndx, mdp, top files automatically generated by Winmostar to analyze the change in distance between specific atoms or groups of atoms. mdp and top files can only be generated when using generic forcefields such as GAFF.
See Atom/Group Distance Change for details.
6.13.11.19. viscosity
The viscosity is calculated from the stress autocorrelation function obtained with the Multiple tau correlator (fix ave/correlate/long). The upper graph shows the normalized autocorrelation function C(t)/C(0) in a log-linear plot, the middle graph shows it in a log-log plot, and the lower graph shows the viscosity obtained from the integrated autocorrelation function. :guilabel:If Fit to ACF & estimate viscosity is checked, a function fitted to the following equation is displayed in green on the autocorrelation function plot.
In the lower graph, the short-time side has a large error due to insufficient integration range, while the long-time side has a large error due to insufficient sampling of the autocorrelation function.
Also, the time (excluding the short-time oscillation component) that the fitted function is below the value entered for Threshold for switching integration from raw to fitted is displayed in the Switch integration at. The Estimated Viscosity displays the viscosity obtained by integrating the autocorrelation function with the trapezoidal formula. The numerical integration is performed over the Switch integration at time, with the short integration over the autocorrelation function obtained directly from the calculation and the long integration over the function fitted to the above equation. If the fit in the above equation is good, the effect of sampling error can be reduced by integrating the function fitted to the long side in the above equation. If the fit is not good, the viscosity can be read directly from the values in the lower graph.
If Plot Autocorrelation Function is not checked, only the viscosity obtained from the integrated value of the autocorrelation function will be plotted. This is useful when determining the interval mean from the graph Options.
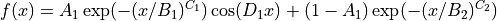
6.13.11.20. Thermal Conductivity
Calculate thermal conductivity from the autocorrelation function of the heat flow obtained with the multiple tau correlator (fix ave/correlate/long). The upper graph shows the normalized autocorrelation function C(t)/C(0) in a log-log plot, the middle graph shows a log-log plot, and the lower graph shows the thermal conductivity obtained from the integrated value of the autocorrelation function. The autocorrelation function plot shows the function fitted to the lower equation in green.
In the lower graph, the short-time side has a large error due to insufficient integration range, while the long-time side has a large error due to insufficient sampling of the autocorrelation function.
Also, the time (excluding the short-time oscillation component) that the fitted function is below the value entered for Threshold for switching integration from raw to fitted is displayed in the Switch integration at. The Estimated Viscosity displays the thermal conductivity obtained by integrating the autocorrelation function with the trapezoidal formula. The numerical integration is performed over the Switch integration at time, where the short time side is integrated over the autocorrelation function directly obtained from the calculation, and the long time side is integrated over the function fitted to the above equation. Integrating the long time side over the function fitted to the above equation reduces the effect of errors due to under-sampling.
6.13.12. Dissipative Particle Dynamics
6.13.12.1. DPD Cell Builder
Create a simulation cell for dissipative particle dynamics.
- Reset
Restore all settings to default.
- Monomers Available
Select the monomer (particle) that constitutes the polymer chain.
- >> Add >>
Add selected monomers.
- << Delete <<
Delete the added monomer.
- Branch
- Start
Specify the branch start position.
- End
Specify the branch end position.
- Monomers Used
A list of added monomer species and number is displayed.
- Clear
Delete all listed monomer species.
- >> Add >>
Add the listed polymer chain to the calculation target.
- << Delete <<
Delete the added polymer chain.
- Export
Output the contents of Monomer Used to a file.
- Import
Reads the contents of Monomer Used from a file.
- Polymers Used
The composition and number of added polymer chains are listed.
- Build
Enter dimensionless densities and build simulation cells.
- Close
Close the window.
6.13.12.2. DPD Potential Editor
Winmostar Create and edit a potential file for proprietary dissipative particle dynamics.
- Potential Files
Select the potential file used for dissipative particle dynamics.
- New
We will create a new potential file.
- Delete
Delete the selected potential file.
- Mass tab
- Species
The name of the monomer (particle) is displayed.
- Mass
Set the mass (dimensionless).
- Bond tab
- R_0
Set bond (bond) potential parameter R_0 (equilibrium distance, dimensionless).
- TO
Set bond (bond) potential parameter K (spring constant, dimensionless).
- Nonbond tab
- Aij
Enter the unbonded potential parameter Aij (dimensionless).
- Rcut
Enter the unbonded potential parameter Rcut (cutoff radius, dimensionless).
- Gamma
Enter the coefficient of friction (dimensionless).
- Set
The set potential parameters are reflected in the list.
- OK
Save the set potential parameters in the potential file and close the window.
- Close
Discard the settings and close the window.