6.12. メニュー¶
LAMMPSに関するメニューです。
LAMMPSをインストールする方法は インストール に記載しています。
6.12.1. 力場を割り当て¶
力場を設定します。ソルバの種類に応じて、選択肢が変化します。
自動で割り当てられるパラメータをテキストファイル上で編集してから計算したい場合は、まず 自動でパラメータを割り当て で一旦パラメータを割り当て、その後の キーワード設定 ウィンドウの Options タブで Dump all files for remote をクリックしファイルを保存してください。その後、そのファイルを編集し、編集後のファイルをWinmostarで開いてください。そして、再度 力場を割り当て 機能を起動し、 トポロジファイルに書かれたパラメータを使用 または メインウィンドウのファイルに書かれたパラメータを使用 を選択してください。
- 自動でパラメータを割り当て
新たに力場パラメータを割り当てます。
- (一般)
タンパク質、水分子以外の分子の力場を指定します。 内部では、GAFF, GAFF2, OPLS/AA-L+GAFFの場合は acpype 、Dreidingの場合は内製プログラム、UFFの場合はOpenBabelを独自に拡張したプログラムが使用されます。 Dreidingの設定は
polymer/dreiding.lib.txtに書かれています。 UFFの詳細は Universal Force Field を確認してください。- Exception
特定の分子に対し、(General)にて選択した力場を使わず、ユーザが指定するLJパラメータを割り当てます。 サブウィンドウの左の欄にてLJパラメータを指定したい分子にチェックを入れ、右の欄でLJパラメータを入力します。
注釈
例えば固液界面系において固相の原子にLJパラメータを割り振りたい時などに使用します。
- (タンパク質/イオン)
タンパク質、単原子の力場を指定します。ここで、PDBやgroフォーマットにおいてアミノ酸残基の名前が割り当てられている原子がタンパク質として認識されます。内部的には gmx pdb2gmx が使用されます。
警告
残基名を含まないファイルから分子構造を読み込んだ場合は本機能を使用できません。
- (水分子)
- 水分子の力場を指定します。 溶媒を配置/セルを構築 で選択した水モデルを指定する必要があります。内部的にはCygwinにインストールされたGromacsのトポロジのライブラリからパラメータを取得します。
- タンパク質向けに[position_restraints]を追加
- タンパク質が存在する場合は Advanced タブにおける -POSRES で位置を拘束するための情報(
[position_restraints]セクション)をトポロジファイルに書き込みます。タンパク質が存在しない場合は無視されます。 - 選択原子向けに[position_restraints]を追加
- ユーザが指定する分子に対し、 Advanced タブにおける -POSRES で位置を拘束するための情報(
[position_restraints]セクション)をトポロジファイルに書き込みます。 例えば固液界面系に於いて固相を固定する場合などに使用します。 - 選択原子向けに[distance/angle/dihedral_restraints]を追加
- ユーザが指定する分子に対し、 Advanced タブにおける -POSRES で距離・角度・二面角を拘束するための情報をトポロジファイルに書き込みます。
- Dump Now
現在の設定に基づき、力場が割り当てられたファイルを生成します。
注釈
- 力場の情報をテキストエディタ等で編集してカスタマイズしたい場合は、まず本機能を使用してファイルを保存し、Gromacsの場合はtop、LAMMPSの場合はdataファイルをテキストエディタ等で編集してください。
- 次に、Gromacsの場合は、 トポロジファイルに書かれたパラメータを使用 を選択して OK ボタンをクリックしてください。そして、topファイルの場所を聞かれるので、先ほど保存・編集したtopファイルを開いてください。
- LAMMPSの場合は、 メインウィンドウのファイルに書かれたパラメータを使用 を選択し Next > ボタンをクリックしてください。そして、 力場の種類を選択してください と出るので、使用する汎用力場の種類を選択して OK ボタンをクリックしてください。
- 電荷はメインウィンドウに表示されている構造から取得されます。メインウィンドウに複数種類の電荷が設定されている場合は(例えばGAMESSのログファイルを開きMulliken電荷とLowdin電荷が設定されている場合など)、(高優先)User電荷>NBO電荷>Lowdin電荷>ESP電荷>Mulliken電荷(低優先)の順番に優先され使用されます。
- パラメータファイルを使用(無機物、ReaxFF、DPD向け)
- (LAMMPS向け)無機物用ポテンシャル、ReaxFFまたはDPDを使用したい場合に選択します。 Next > ボタンを押した後に、実際に使用する力場の種類を指定します。
- トポロジファイルに書かれたパラメータを使用
- (Gromacs向け)既に存在しているtopファイルを用いてMD計算を実行する場合に選択します。メインウィンドウには対応するgroファイルを開いておく必要があります。
- メインウィンドウのファイルに書かれたパラメータを使用
- (LAMMPS向け)既に存在しているdataファイルを用いてMD計算を実行する場合に選択します。メインウィンドウには使用するdataファイルを開いておく必要があります。 Next > ボタンを押した後に、使用する力場の種類を指定します。
6.12.2. キーワード設定¶
LAMMPSの計算条件を設定します。設定後、すぐに計算を実行する場合は Run ボタン、一旦メインウィンドウに戻る場合は OK ボタンを押してください。
Run をクリックしたときの挙動は LAMMPS実行 を参照してください。
電荷が割り当てられていない分子がある場合は、 自動で電荷を割り当て が自動で立ち上がります。 力場が割り当てられていない場合は、 力場を割り当て が自動で立ち上がります。
Reset ボタンでデフォルトの状態に戻ります。 Save ボタンでForce Fieldを除く設定を保存します。 Load ボタンで Save にて保存した設定を読み込みます。
- Extending Simulation
継続ジョブを実行します。
詳細は LAMMPS実行 を参照してください。
- Preset
計算条件のプリセットを指定します。各プリセットは以下のキーワードを変更します。
Pair style lj/cut/coul/long lj/cut/coul/long lj/cut/coul/long lj/cut/coul/long Time step 2.0 2.0 2.0 # of time steps 5000 5000 5000 5000 Ensemble minimize nvt npt nve True False False Temperature 300 300 Pressure 1.0 Boundary Condition p p p p p p p p p p p p Reset COM motion linear linear linear linear Tchain 3 3 Pchain 3 Shake tolerance 1e-5 1e-5 1e-5 100 100 100 100 100 100 100 100 Log interval 10 10 10 10 Cutoff (vdW)
Cutoff (Coulomb)
PPPM order 4 4 4 4 K-space accuracy 1e-5 1e-5 1e-5 1e-5
Pair style lj/cut/coul/long lj/cut/coul/long lj/cut/coul/long lj/cut/coul/long Time step 0.5 0.5 0.5 # of time steps 20000 20000 20000 20000 Ensemble minimize nvt npt nve True False False Temperature 300 300 Pressure 1.0 Boundary Condition p p p p p p p p p p p p Reset COM motion linear linear linear linear Tchain 1 1 Pchain 1 Shake tolerance 1e-9 1e-9 1e-9 400 400 400 400 400 400 400 400 Log interval 40 40 40 40 Cutoff (vdW)
Cutoff (Coulomb)
PPPM order K-space accuracy
Pair style lj/cut/coul/cut lj/cut/coul/cut lj/cut/coul/cut lj/cut/coul/cut Time step 2.0 2.0 2.0 # of time steps 5000 5000 5000 5000 Ensemble minimize nvt npt nve True False False Temperature 300 300 Pressure 1.0 Boundary Condition f f f f f f f f f f f f Reset COM motion angular angular angular angular Tchain 3 3 Pchain 3 Shake tolerance 1e-5 1e-5 1e-5 100 100 100 100 100 100 100 100 Log interval 10 10 10 10 Cutoff (vdW)
Cutoff (Coulomb)
PPPM order K-space accuracy
Pair style lj/cut/coul/cut lj/cut/coul/cut lj/cut/coul/cut lj/cut/coul/cut Time step 0.5 0.5 0.5 # of time steps 20000 20000 20000 20000 Ensemble minimize nvt npt nve True False False Temperature 300 300 Pressure 1.0 Boundary Condition f f f f f f f f f f f f Reset COM motion angular angular angular angular Tchain 1 1 Pchain 1 Shake tolerance 1e-9 1e-9 1e-9 400 400 400 400 400 400 400 400 Log interval 40 40 40 40 Cutoff (vdW)
Cutoff (Coulomb)
PPPM order 6 6 6 6 K-space accuracy 1e-9 1e-9 1e-9 1e-9
Pair style reax/c reax/c reax/c reax/c Time step 0.5 0.5 0.5 # of time steps 20000 20000 20000 20000 Ensemble minimize nvt npt nve True False False Temperature 300 300 Pressure 1.0 Boundary Condition p p p p p p p p p p p p Reset COM motion linear linear linear linear Tchain 1 1 Pchain 1 Shake tolerance 1e-9 1e-9 1e-9 400 400 400 400 400 400 400 400 Log interval 40 40 40 40 Cutoff (vdW)
Cutoff (Coulomb)
PPPM order K-space accuracy - MPI
- MPI並列数を指定します。
- Basic
- Units
単位系を指定します。
- real
- 主に分子系で指定します(A, fs, Kcal/mol)。
- metal
- 主に結晶系で指定します(A, ps, eV)。
- lj
- 主にDPD計算で指定します(無次元単位)。
- Atom Style
- 計算する系の種類を指定します。 Units に応じて変化します。
- Pair Style
- 相互作用計算の方法を選択します。
- Force Field/Potential File
Units がrealの場合には、力場の種類を指定します。special_bonds, bond_style, angle_style, dihedral_style, improper_styleキーワードに影響します。
Units がreal以外の場合には、ポテンシャルファイルを選択します。LAMMPS本体をインストールしたフォルダ直下の
Potentialフォルダ内のファイルをリストアップします。選択肢は Units および Pair Style に応じて変わります。- Time Step
- 時間積分の刻み幅を指定します。単位は選択した Unit により変わります。
- # of Time Steps
- 時間積分ステップの最大数を指定します。
- Ensemble
- 時間積分の種類を指定します。
nvt(温度一定のカノニカルアンサンブル),npt(温度、圧力一定のアンサンブル),nve(体積とエネルギー一定のミクロカノニカルアンサンブル),minimize(CG法によるエネルギー最小化)のいずれかを選択します。- Generate Velocity
- チェックをした場合は初速度が与えられます。
- Random Seed
- 初速度発生時の擬似乱数の種を指定します。
- Temperature
- 目標温度を指定します。アニーリング計算時には始状態の温度を指定します。
- Tdamp
- 温度制御の時定数パラメータを指定します。
- Pressure Control
- 圧力制御の際のセルの動かし方を指定します。
- Pressure
- 目標圧力を指定します。
- Pdamp
- 圧力制御の時定数パラメータを指定します。
- Advanced
- Boundary X Y Z
- 周期境界条件を指定します。
p(periodic),f(non-periodic and fixed),s(non-periodic and shrink-wrapped),m(non-periodic and shrink-wrapped with a minimum value)のいずれかを選択します。- Energy Tolerance
- minimize計算時のエネルギーに関する打ち切り誤差を指定します。
- Force Tolerance
- minimize計算時の力に関する打ち切り誤差を指定します。
- Reset COM Motion
- MD計算時に系全体の重心の運動を凍結する方法を選びます。
- Reset Interval
- Reset COM Motion の頻度をタイムステップで指定します
- Tchain
- Nose-Hoover chainの段数を指定します。
- Pchain
- 圧力制御の段数を指定します。
- Constrain hydrogen atoms
- 水素原子をSHAKE法で拘束します。
- SHAKE tolerance
- SHAKE法の打ち切り誤差を指定します。
- Automatically disable Shake if CH4-like molecule exists
- メタン状の分子が含まれている時に自動でSHAKE法を無効にします。
- Calculate as rigid body
- 分子を剛体として扱います。
- Set "box tilt large"
- シミュレーションセルの変形の許容度合を指定します。
- Output
- Dump Interval (dump)
- dump形式で座標を出力する頻度をタイムステップ数で指定します。
- Dump Interval (xtc)
- xtc形式で座標を出力する頻度をタイムステップ数で指定します。
- Dump Interval (xyz)
- xyz形式で座標を出力する頻度をタイムステップ数で指定します。
- Log Interval
- log ファイルにエネルギー変数を書き出す頻度をタイムステップ数で指定します。
- Print log in high precision
- log ファイルに書き出すエネルギー変数の桁数を増やします。
- Sort dump file by id
- dumpファイル内の粒子の並びをid(通し番号)でソートされた形にします。
- Calculate Fluctuation Properties
- 熱力学量の揺らぎから比熱と等温圧縮率をon-the-flyで計算し出力します。
- Calculate Thermal Conductivity
- 原子の流速の自己相関関数とGreen-Kubo式から熱伝導率をon-the-flyで計算し出力します。
- Calc Interval
- 熱伝導率計算における自己相関関数の算出頻度を指定します。
- ACF Length
- 熱伝導率計算における自己相関関数の長さを指定します。
- Calculate viscosity
- 圧力テンソルの自己相関関数とGreen-Kubo式から粘度をon-the-flyで計算し出力します。
- Calc Interval
- 粘度計算における自己相関関数の算出頻度を指定します。
- ACF Length
- 粘度計算における自己相関関数の長さを指定します。
- Interaction
- Modify cutoff radii not to exceed L/2
- チェックを入れた場合は、Cutoff (vdw), Cutoff (Coulomb)が格子定数の半分を超えないように自動調整します。
- Neighbor Search
- 近接粒子探索時のアルゴリズムを指定します。
- Neighbor Skin
- 近接粒子探索時の探索半径の余分を指定します。
- Cutoff(vdw)
- vdw(LJ)ポテンシャルのカットオフ半径を指定します。
- Enable Long Range Correction
- vdwポテンシャルのカットオフ補正項の有無を指定します。
- Cutoff(Coulomb)
- Coulomb(静電)ポテンシャルのカットオフ半径を指定します。
- Disable Ewald(PPPM) if no charge exists
- 系が電荷を持たない場合に、自動でEwald(PPPM)法を無効にします。
- Automatically set Nmesh
- Pair Style =
lj/cut/coul/longの際に使用されるPPPM法のメッシュ数をK-space accuracyから自動的に設定します。- Nmesh for kx, ky, kz
- PPPM法のメッシュ数を指定します。
- PPPM Order
- PPPM法のSpline補間次数を指定します。
- K-space accuracy
- PPPM法の許容相対誤差を指定します。
- Non-equiliibrium (1)
- Enable Elongation
- 伸長計算を有効にします。 Ensemble が
minimize以外の時に指定できます。- Affine Transformation
- 伸長計算時に原子位置をシミュレーションセルに合わせてアフィン(相似)変形するか指定します。
- Eng. Strain Rate
- 伸長計算時の伸長速度を工業ひずみで指定します。 Max Eng. Strain には最終ステップにおけるひずみの予測値が表示されます。
- Preserve Volume
- 伸長計算時に、シミュレーションセルの体積を一定に保つよう伸長方向に垂直な方向のセルサイズを変形させます。
- Enable Pulling
- 指定した原子群を一定速度で移動させるPull計算を有効にします。 Ensemble が
minimize以外の時に指定できます。- Pulled Atoms
- 予め 登録グループを選択 でPullしたい原子を登録しておいた状態で Select Group ボタンを押し、そのグループを選択します。
- Pull Velocity
- Pull計算時の、Pull速度を指定します。
- Enable Simulated Annealing
- アニーリング計算(温度を一定速度で変化させる計算)を有効にします。 Ensemble が
nvt,nptの時に指定できます。 Temperature の値が始状態の温度、 Final Temperature の値が終状態の温度となります。- Final Temperature
- アニーリング計算時の終状態の温度を指定します。
- Annealing Rate
- アニーリング計算時の加熱または冷却速度が表示されます。
- Enable pressurization
- 圧縮計算(圧力を一定速度で変化させる計算)を有効にします。 Ensemble が
npt,nphの時に指定できます。 Pressure の値が始状態の圧力、 Final Pressure の値が終状態の圧力となります。- Final Pressure
- 圧縮計算時の終状態の圧力を指定します。
- Enable electric field
- 外部電場を与えます。 Sine wave を選ぶと、正弦波的に電場を与えます。 Constant を選ぶと、定常的に電場を与えます。
- Amp & Freq
各方向の強度(Amp)と周波数(Freq)を与えます。 Enable electric field で Sine wave を選んだ時は下式で電場が与えられ、Aが強度、fが周波数となります。 Constant を選んだ時は強度のみが使われます。
- Restraint
- Enable Restraint
- 指定した2原子間の距離を拘束した計算を実施します。Ensemble が
minimize以外の時に指定できます。- Restrained Atoms
- Set ボタンをクリックすると、マーカーが付いた2原子が拘束のターゲットとなります。
- Bond Length
- 拘束計算時の、2原子間の拘束距離を指定します。
- Initial Strength
- 拘束計算時の、始状態における拘束ポテンシャルのバネ係数を指定します。
- Final Strength
- 拘束計算時の、終状態における拘束ポテンシャルのバネ係数を指定します。
- Enable Position Restraint
- 指定した原子の絶対座標を固定した計算を実施します。固定されていない原子の温度はlogにTempFreeとして出力されます。
- Restrained Atoms
- 予め 登録グループを選択 で拘束したい原子を登録しておいた状態で Select Group ボタンを押し、そのグループを選択します。
- Use spring potential
- Restrained Atoms に指定した原子を初期位置からのばねポテンシャルで拘束します。 Reset positions of restrained atoms after run にチェックが入った場合は、計算終了後は初期位置に戻されるため、Extending simulationを繰り返しても最初に指定した位置から離れて移動することはありません。
- Spring constant
- ばねポテンシャルを使う際のばね係数を指定します。
- Reset positions of restrained atoms after run
- ばねポテンシャルを使う際、計算終了後に拘束した原子の位置を初期位置に戻します。
- Automatic
- Rescale velocities to..
- NVEアンサンブルにおいて目標温度に系の温度を近づけたい時に使います。計算中の平均温度とここで入力した温度からスケーリング係数を算出して、最終構造の各粒子の速度をスケーリングします。
- Rescale box size to..
- NPTアンサンブルで計算した後に、設定圧力に近い状態でNVEまたはNVTアンサンブルで計算した場合に使用します。最終構造を、計算中の平均セルサイズにスケーリングします。
- Manual entry
- 生成されるLAMMPSのインプットスクリプト(inファイル)の中身が表示されます。この場所で直接編集することも可能です。
- Options
- Make a Backup of Working Directory
- 作業ディレクトリのバックアップを行う際に選択します。
- Restore Working Directory
- 継続ジョブが異常終了時など、作業ディレクトリを実行前の状態に戻す際にクリックします。
- Dump all files for remote
- Linux環境でのジョブ実行に必要なファイルを出力します。 リモートジョブ 機能で生成されるファイルと同じファイルが出力されます。
- Generate gro & ndx files every time
- チェックが入っていない場合は、継続ジョブの時にgroとndxファイルを生成しません。
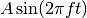
6.12.3. LAMMPS実行¶
LAMMPSを実行します。 状況に応じて実行方法が異なります。
- (デフォルト) Extending Simulation にチェックがなく、 力場を割り当て において 自動でパラメータを割り当て または パラメータファイルを使用(無機物、ReaxFF、DPD向け) を選択した場合
- dataファイル(座標とトポロジを含むファイル)を新規に生成してからジョブを開始します。
- Extending Simulation にチェックがなく、 力場を割り当て において メインウィンドウのファイルに書かれたパラメータを使用 を選択した場合
- メインウィンドウで開かれているdataファイルを使用してジョブを開始します。
- Extending Simulation にチェックがある場合
- メインウィンドウで開かれているdataファイルに紐づけられた作業ディレクトリの中にある
lmp_tmp_final.data用いてジョブを開始します。実行に伴い以下のファイルが生成されます。 例として入力ファイルが
water.dataの時のファイル/フォルダ名を併記しています。
種類 説明 water.logLAMMPSのログファイルです。 water.batwater_lmp_tmp\作業ディレクトリには以下のファイルが生成されます。 ここでは主要なファイルのみ示しています。
種類 説明 lmp_tmp.datalmp_tmp.inlmp_tmp.logwater.logと同じものです。lmp_tmp.dumplmp_tmp.restartlmp_tmp_final.datapostproc.shlmp_tmp_final.dataが、そのままではLAMMPSの実行には不十分なため、不十分な情報を補うための処理を行うスクリプトです。lmp_tmp.xtclmp_tmp.xtclmp_tmp.groヒント
作業ディレクトリ
作業ディレクトリとは、メインウィンドウで開かれているファイルの名前に接尾辞を加えた名前のフォルダです。
- 接尾辞はソルバの種類により変わります。
- 例えばGromacsの場合は、メインウィンドウで開かれているファイルが
aaa.groで、接尾辞が_gmx_tmpの場合、作業ディレクトリの名前はaaa_gmx_tmpとなります。メインウィンドウで開かれているファイルと同じ階層に置かれている必要があります。
継続ジョブの時も同名の作業ディレクトリで処理が流れますが、デフォルトでは継続ジョブの実施直前に、直前のジョブの作業ディレクトリのバックアップが作成されます。
- バックアップの名前は、重複する名前が存在しない範囲で一番小さい番号が付いたものになります。例えば、作業ディレクトリが
aaa_gmx_tmpのときはaaa_gmx_tmp1となります。- 番号が付いていないディレクトリが常に最新のものとなります。
ジョブは Winmostar Job Manager を通じて実行されます。
6.12.4. ログを表示¶
LAMMPSのログファイル(*.log)をテキストエディタで開きます。
6.12.5. アニメーション¶
dataファイルとdumpファイルを選択し、MD計算のトラジェクトリをアニメーション表示します。
メインウィンドウのファイル名は変化しません。
アニメーション表示の操作方法は Animationウィンドウ を参照してください。
6.12.6. エネルギー変化¶
ログファイルを選択し、エネルギー、温度、圧力などの各種熱力学量のグラフを表示します。thermo_styleで指定した値をプロットすることができます。
サブウィンドウの操作方法は Energy Plotウィンドウ を参照してください。
6.12.7. 最終構造を読み込み¶
*_lmp_tmp\lmp_tmp_final.groを開きます。本機能を使うとメインウィンドウのファイル名は変化しません。
6.12.8. 結果解析¶
6.12.8.1. 動径分布関数¶
LAMMPSが出力したxtcファイルとWinmostarが自動生成したgro, ndxファイルを選択し、動径分布関数を表示します。
詳細は 動径分布関数 を参照してください。
6.12.8.2. 自己拡散係数/平均二乗変位¶
LAMMPSが出力したxtcファイルとWinmostarが自動生成したgro, ndxファイルを選択し、平均二乗変位と自己拡散係数を表示します。
詳細は 自己拡散係数/平均二乗変位 を参照してください。
6.12.8.4. 比誘電率/双極子モーメント¶
LAMMPSが出力したxtcファイルとWinmostarが自動生成したgro, ndx, mdp, topファイルを選択し、散乱関数を表示します。mdp, topファイルはGAFFなどの汎用力場使用時のみ生成可能なため、パラメータファイルの使用時などは本機能を利用できません。
詳細は 比誘電率/双極子モーメント を参照してください。
6.12.8.5. 密度分布¶
LAMMPSが出力したxtcファイルとWinmostarが自動生成したgro, ndx, mdp, topファイルを選択し、密度分布を表示します。mdp, topファイルはGAFFなどの汎用力場使用時のみ生成可能なため、パラメータファイルの使用時などは本機能を利用できません。
詳細は 密度分布 を参照してください。
6.12.8.6. 自由体積¶
LAMMPSが出力したxtcファイルとWinmostarが自動生成したgro, ndx, mdp, topファイルを選択し、自由体積を表示します。mdp, topファイルはGAFFなどの汎用力場使用時のみ生成可能なため、パラメータファイルの使用時などは本機能を利用できません。
詳細は 自由体積 を参照してください。
6.12.8.7. 各種自己相関関数¶
Green-Kubo式を用いた熱伝導率、粘度算出時に出力される自己相関関数を表示します。
6.12.8.8. 距離/角度/二面角分布¶
LAMMPSが出力したxtcファイルとWinmostarが自動生成したgro, ndx, mdp, topファイルを選択し、選択グループ間の距離、角度、または二面角の分布を表示します。mdp, topファイルはGAFFなどの汎用力場使用時のみ生成可能なため、パラメータファイルの使用時などは本機能を利用できません。
詳細は 距離/角度/二面角分布 を参照してください。
6.12.9. 散逸粒子動力学¶
6.12.9.1. DPDセルビルダ¶
散逸粒子動力学用のシミュレーションセルを作成します。
- Monomers Available
- ポリマー鎖を構成するモノマー(粒子)を選択します。
- # of Monomers
- 選択したモノマーの数を指定します。
- >> Add >>
- 選択したモノマーを追加します。
- Branch
- Start
- 分岐開始位置を指定します。
- End
- 分岐終了位置を指定します。
- Monomers Used
- 追加したモノマー種と数がリスト表示されます。
- Clear
- リストアップされたモノマー種を全て削除します。
- << Delete <<
- 追加したモノマーを削除します。
- # of Polymers
- ポリマー鎖の数を指定します。
- >> Add >>
- リストアップされたポリマー鎖を計算対象に追加します。
- Polymers Used
- 追加したポリマー鎖の構成と本数がリスト表示されます。
- << Delete <<
- 追加したポリマー鎖を削除します。
- Density
- 系の密度(無次元)を指定します。
- Build
- シミュレーションセルを構築します。
- Reset
- すべての設定をデフォルトに戻します。
- Close
- ウィンドウを閉じます。
6.12.9.2. ポテンシャル編集¶
Winmostar独自形式の散逸粒子動力学用のポテンシャルファイルを作成・編集します。
- Potential Files
- 散逸粒子動力学に用いるポテンシャルファイルを選択します。
- New
- 新たにポテンシャルファイルを作成します。
- Delete
- 選択したポテンシャルファイルを削除します。
- Massタブ
- Species
- モノマー(粒子)名が表示されます。
- Mass
- 質量(無次元)を設定します。
- Bondタブ
- R_0
- 結合(ボンド)ポテンシャルパラメータR_0(平衡距離、無次元)を設定します。
- K
- 結合(ボンド)ポテンシャルパラメータK(バネ定数、無次元)を設定します。
- Nonbondタブ
- Aij
- 非結合ポテンシャルパラメータAij(無次元)を入力します。
- Rcut
- 非結合ポテンシャルパラメータRcut(カットオフ半径、無次元)を入力します。
- Set
- 設定したポテンシャルパラメータがリストに反映されます。
- OK
- 設定したポテンシャルパラメ-タをポテンシャルファイルに保存してウィンドウを閉じます。
- Close
- 設定内容を破棄してウィンドウを閉じます。