6.16. メニュー
Quantum ESPRESSOに関するメニューです。
6.16.1. Quantum ESPERSSOの設定方法
Quantum ESPRESSOをインストールするには、2023/04/05バージョン以降のCygwinWMをインストールしてください。CygwinWMの
/opt_win/QuantumESPRESSO*/bin/にpw.exeなどが収められています。WinmostarからQuantum ESPRESSOを利用するための設定は ツール ‣ 環境設定 メニュー で行います。Winmostar V11.5.0以降を新規インストールしCygwinWM 2023/04/05バージョン以降を利用する場合は設定不要です。
まず で使用するQuantum ESPRESSOの
pw.exeを選択します。次に、 で MPICH を選択するか、 Select を選択し使用するMPIのmpiexec.exeを選択します。CygwinWMに収められたQuantum ESPRESSOを使う場合は、CygwinWMの下の/opt_win/MSMPI/Bin/mpiexec.exeを選択します。次に、 で mpiexec.exe の引数を入力します。CygwinWMに収められたQuantum ESPRESSOを使う場合は、-np %WM_NUM_PROC% と入力します。最後に、 で選択したpw.exeのバージョンを選択します。CygwinWMに収められたQuantum ESPRESSOを使う場合は、 7.1 を選択します。擬ポテンシャルファイルを追加したい場合は、 ツール ‣ 環境設定 メニュー をクリックし、開いたフォルダに擬ポテンシャルファイルを追加します。擬ポテンシャルファイルの拡張子はupf(大文字、小文字の区別なし)である必要があります。擬ポテンシャルフォルダを新規に作成し選択することも可能です。
RISM計算用のMOLファイルを追加したい場合は、 ツール ‣ 環境設定 メニュー をクリックし、開いたフォルダにMOLファイルを追加します。擬ポテンシャルファイルの拡張子はmol(大文字、小文字の区別なし)である必要があります。MOLファイル用フォルダを新規に作成し選択することも可能です。
リモートマシンにQuantum ESPRESSOをインストールする方法は インストール に記載しています。
6.16.2. ワークフロー設定
プロジェクトモードにおけるQuantum ESPRESSOの計算フローを設定、実行します。
- Preset
設定のプリセットを呼び出し、保存します。
- # of Jobs
ジョブの数を指定します。
- Enable parameter/structure scan
この機能を使うためにはアドオンの購入が必要です。特定のパラメータだけが異なる複数の計算を流したり(パラメータスキャン)、複数の構造に対し同一のパラメータで計算を流す(構造スキャン)ことが可能となります。
Config をクリックするとスキャン計算の設定画面が出現します。
パラメータスキャンの際は Target Variable に%WM_SCAN1%を選択し、 Values の各行に%WM_SCAN1%に設定したいパラメータを入力します。そして、ワークフロー設定ウィンドウまたはキーワード設定ウィンドウにおいて設定したいパラメータに%WM_SCAN1%と入力します。Valuesの各行にスペース区切りで値が記入されている場合は、1列目の値を%WM_SCAN1_1%、2列目の値を%WM_SCAN1_2%...といった具合に参照することができます。
並列数をスキャンしたい場合は、# of Threadsまたは# of Procsを選択します。
構造スキャンの際は、分子表示エリアでアニメーションが出現した状態(SDFファイルを開くなど)で、 Target Variable に%WM_STRUCT%を選択します。また、構造スキャン実行時には作業フォルダにlabel_scan_struct.txtというファイル名で、アニメーション表示エリアのリストの文字列が保存されます。
スキャン計算の終了後は を利用して計算結果を集計します。
- Import
Exportで出力した設定を読み込みます。ボタン右の矢印をクリックすると、過去同じプロジェクトまたはWinmostar上で使用した設定を呼び出すことができます。
- Export
設定をファイルに出力します。
- OK
設定した内容で計算を実行またはファイルを生成します。詳しくは プロジェクトモードの場合 を参照してください。
- Details
計算条件を詳細に設定します。 キーワード設定 が立ち上がります。
- Task
計算の種類を指定します。
- Charge
系全体の電荷を指定します。
- # of bands
バンド数を指定します。
- Spin
スピン分極計算の設定をします。Polarized(Manual)ではUse Spin Density asがFalseとなり各元素が1種類の初期磁化モーメントしか持てないため、磁性体の計算ではPolarizedの方が柔軟に初期磁化モーメントを設定できます。
- Cutoff energy
波動関数のカットオフエネルギー(ecutwfc)を設定します。
- Manually specify cutoff energy
チェックが入っていない場合はPrecisionからCutoff energyを自動指定します。
- K points
K点の計算方法を指定します。
- Pressure
圧力を指定します。
- Phonon(DFPT)
DFPT(ph.x)を用いたフォノン計算の設定をします。
- Use Bravais-lattice index
チェックを入れた場合は入力ファイルをibrav=0以外で作成します。バンド構造、フォノンバンドの計算時にibravから特殊点を設定する必要があるのでチェックを入れてください。
- Pseudopotential
擬ポテンシャルファイルを指定します。Typeを変更するとFunctionalとPseudo fileが絞り込まれます。Functionalを変更するとPseudo fileが絞り込まれます。所望の選択肢が出現しない場合の操作方法は キーワード設定 のPseudopotentialを参照してください。
- Properties
計算時に合わせてチェックを入れた物性を計算します。
- Precision
計算精度を設定します。
- Metal
スメアリングを有効にします。(occupations=smearing)また、Precision(上記)の内容も調整します。
6.16.3. キーワード設定
Quantum ESPRESSOの計算条件を設定します。設定後、すぐに計算を実行する場合は Run ボタン、一旦メインウィンドウに戻る場合は OK ボタンを押してください。
Run をクリックしたときの挙動は 実行 を参照してください。
Reset ボタンでデフォルトの状態に戻ります。 Save ボタンでForce Fieldを除く設定を保存します。 Load ボタンで Save にて保存した設定を読み込みます。
キーワード表示エリア に表示されているQuantum ESPRESSOのキーワードと本機能のウィンドウに設定された内容が異なる場合は、キーワード表示エリアからキーワードを読み込むか尋ねられます。
本機能を呼び出すときに、メインウィンドウに表示された構造がプリミティブセルに変化できる場合は、自動で 格子を変換 (Primitive-Conventional) が実行されます。
- Output Directory
データの出力先フォルダ(outdir)を指定すると同時に、ジョブの新規・継続実行を指定します。
- Create
新規にデータの出力先フォルダを作成します。outdirは新規フォルダに設定されます。
- Continue
メイン画面に読み込まれている、直前に実行されたQEのジョブを継続します。outdirは直前に実行されたジョブのoutdirに設定されます。
- Select
開いたダイアログで指定したフォルダを出力先に指定し、そのフォルダのデータからジョブを継続します。outdirはここで指定したものに設定されます。
- Preset
設定のプリセットを選択します。各プリセットは以下のキーワードを変更します。
scf
scf
scf
relax
vc-relax
md
cp
scf
False
True
True
False
False
False
False
False
gamma
gamma
gamma
gamma
gamma
False
False
False
False
False
True
False
False
False
True
True
False
False
False
False
False
smearing
bfgs
bfgs
verlet
none
bfgs
False
False
False
True
True
True
True
True
False
False
False
False
True
False
False
True
False
False
False
False
True
False
False
False
second_order
second_order
sd
angstrom
angstrom
angstrom
angstrom
angstrom
crystal
False
True
True
False
False
False
False
False
False
True
True
False
False
False
False
False
False
True
False
False
False
False
False
False
False
False
True
False
False
False
False
False
- Use MPI
QEの実行時にMPIを用いて並列計算を実行するか指定します。横の欄にはMPIプロセス数を入力します。
- Basicタブ
- restart_mode
from_scratchの時は、過去のジョブの情報を使わずにジョブを開始します。restartの時はoutdirの情報から引き継いでジョブを開始します。Automatically setの時は、プロジェクトモードでは新規ジョブでfrom_scratch、それ以外でrestartとなります。ファイルモードでは、Output Directory=Createでfrom_scracth、それ以外でrestartとなります。
- calculation
計算の種類を選択します。
- # of bands
括弧内に選択されたは擬ポテンシャルを使用したときの価電子数が表示されます。価電子数の自動計算に失敗した場合はN/Aと表示されます。
- Do not specify
nbndが自動で設定されます。
- Specify nbnd
明示的にバンド数を指定します。
- Specify nbnd(Relative)
価電子バンド数からの相対値で指定します。
- Use nbnd
バンド数を指定します。チェックを入れない場合は自動で設定されます。
- K_POINTS
K点の指定方法をプルダウンから選び、下のテキストボックスにQEの書式でK点を指定します。 SeeK-path などのウェブサイトで生成したk点の情報を使いたい場合は、K_POINTSの種類(crystalなど)を選択した後でその下のテキストボックスにk点の座標と点数を貼り付けてください。入力ファイルに書き出されるk点数(nks)は自動的に補われます。
- gamma
Γ点のみで計算されます。
- automatic
Monkhorst-Pack法を使用します。「(kx方向の分割数) (ky方向の分割数) (kz方向の分割数) (kx方向のシフトフラグ) (ky方向のシフトフラグ) (kz方向のシフトフラグ)」(スペース区切り)を入力します。シフトフラグは0のときにシフトなし(k点がΓ点を含む)、1のときにシフトあり(k点がΓ点を跨ぐ)となります。
- automatic(by spacing)
Monkhorst-Pack法を使用します。各方向の分割数はSpacingパラメータ( Spacingパラメータ )で設定されます。
- automatic(by spacing,slab)
Monkhorst-Pack法を使用します。各方向の分割数はSpacingパラメータ( Spacingパラメータ )で設定されます。ただし自動的に検出されるスラブ垂直方向の分割数は1に設定されます。
- その他
詳細はpw.xのマニュアルまたはQEインストールフォルダ以下のdoc/brillouin_zones.pdfをご参照ください。
- Set default k-path
メインウィンドウに表示されている結晶について検出されたブラベー格子(ibrav)のデフォルトのk点パスをUserPref/kpath_default.txtから取得して設定します。
- nosym
空間対称性の利用の有無を指定します。
- noinv
時間反転対称性の利用の有無を指定します。
- Set ibrav = ... and celldm
メインウィンドウにプリミティブセルが表示されていて、チェックが入っている場合は、適当なibravとcelldmを設定します。 チェックが入っていない場合は、ibrav=0とし、CELL_PARAMETERSを設定します。
- ecutwfc
波動関数の計算時の平面波のカットオフエネルギーを指定します。
- Ecut for US/PAW
ウルトラソフト、PAWの擬ポテンシャルファイルを選択した際のecutrhoの指定方法を設定します。
- Specify ecutrho/ecutwfc
ecutrhoとecutwfcの比とecutwfcの値からecutrhoを設定します。
- Specify ecutrho
ecutrhoの値を直接設定します。
- ecutrho
電子密度およびポテンシャル計算時の平面波のカットオフエネルギーを指定します。
- tot_charge
シミュレーションセル内の系全体の電荷を指定します。
- occupations
金属の場合はsmearing, DOS計算の場合はtetrahedronを指定します。
- ion_dynamics
イオン(原子核)位置を変化させるアルゴリズムを指定します。
- cell_dynamics
シミュレーションセルを変化させるアルゴリズムを指定します。
- tprnfor
力を計算します。
- tstress
圧力テンソルを計算します。
- Advancedタブ
- conv_thr
SCF計算時のエネルギーの許容誤差を指定します。
- etot_conv_thr
構造最適化(relax)計算時のエネルギーの許容誤差を指定します。
- forc_conv_thr
構造最適化(relax)計算時の力の許容誤差を指定します。
- press_conv_thr
セルの構造最適化(relax-vc)計算時の圧力の許容誤差を指定します。
- electron_maxstep
SCF計算の最大反復数を指定します。
- nstep
構造最適化(relax)計算の最大ステップ数およびMD計算のステップ数を指定します。
- upscale
構造最適化(relax)計算の最中にconv_thrを自動的に小さくする際の係数を指定します。
- diagonalozation
対角化アルゴリズムを指定します。
- diago_david_ndim
Davidsonアルゴリズムでの対角化時のワークスペースの大きさを指定します。
- spline_ps
cubic splineで擬ポテンシャルを内挿します。GIPAW計算で有用です。
- la2F (for pw.x)
pw.xにおいて電子フォノン相互作用計算用に固有値の書かれたファイルを出力します。
- smearing
占有平滑化(smearing)の方法を指定します。
- degauss
占有平滑化のパラメータを指定します。
- mixing_beta
SCF計算における新旧のKS軌道の混合比を指定します。1に近いほど予測値の割合が大きくなります。
- mixing_mode
新旧のKS軌道の混合アルゴリズムを指定します。
- vdw_corr
ファンデルワールス(分散)力の補正方法を指定します。
- Use input_dft
チェックを入れた場合は、汎関数の設定を擬ポテンシャルファイルに書かれた設定に対し上書きします。
- nqx1/2/3
Fock演算子計算時のk点サンプリング数を指定します。Defaultの場合はK_POINTSと同じ値を使います。
- cell_dofree
シミュレーションセルを動かす際の自由度(方向)を指定します。
- Use cell_factor
擬ポテンシャルテーブルの構築パラメータを明示的に指定します。vc-relax(セルサイズの変化を伴う構造最適化計算)の際に大きい値を設定した方がいい場合があります。
- Spin/DFT+Uタブ
- nspin
スピン分極計算の設定をします。
- Use tot_magnetization
チェックを入れた場合は、ここで系全体の磁化を指定します。チェックしない場合はstarting_magnetizationを指定します。
- starting_magnetization
各原子種の磁化の初期値を与えます。この場合同元素の原子がすべて同じ値を持つため、反強磁性体を計算したい場合はUse Spin Density as starting_magnetizationを使用してください。
- noncolin
ノンコリニア計算の有無を指定します。チェックを入れた場合はnspinを入力ファイルに出力しません。nspinは出力されませんが、starting_magnetizationを設定するには同じタブ上でnspin=2に設定しておく必要があります。
- lspinorb
ノンコリニア計算時にスピン軌道相互作用付きの擬ポテンシャルファイルを使えるようにします。
- Use Spin Density as starting_magnetization
メインウィンドウで設定されたSpin Densityの値をそのままstarting_magnetizationとして使用します。チェックを入れた場合は上のstarting_magnetizationが無視されます。反強磁性体のように同元素の原子が異なるstarting_magnetizationを持つ場合はこの機能を使用してください。
- lda_plus_u
LDA+U計算を実行します。
- Hubbard_U/alpha
各原子種のHubbardのUおよびalphaパラメータを指定します。
- Phononタブ
- Run Phonon Calculation as Postprocess
フォノン計算を実行します。 具体的には、pw.xを実行した後にph.xを実行します。 この項目を有効にするためには、Calculationにscf, nscfまたはrelaxを選ぶ必要があります。 SCF計算またはRelax計算が終わった状態からph.xを実行したいときには、Calculation=nscfで実行するとpw.xがすぐに終了しph.xの処理にすぐに入ります。 ph.xなどの入出力ファイルは作業フォルダ内に作成されます。
- tr2_ph
フォノン計算の打ち切り誤差を指定します。
- alpha_mix(1)
mixing factorを指定します。
- epsil=.True.
フォノン計算から得られる誘電率(電子系)を計算します。
- lraman=.True.
Ramanスペクトルの計算を含めます。
- electron_phonon
電子フォノン相互作用計算の方式を指定します。
- el_ph_sigma
電子フォノン相互作用計算中のdouble-deltaスメアリングの間隔を指定します。
- el_ph_nsigma
電子フォノン相互作用計算中のdouble-deltaスメアリングの数を指定します。
- Use fildvscf
電子フォノン相互作用計算で使うファイルをq2r.x実行時に出力します。
- la2F = .true. (for q2r)
電子フォノン相互作用計算でチェックを入れてください。
- asr(for dynmat)
フォノン計算後のポスト処理(dynmat.x)においてAcoustic Sum Ruleの与え方を指定します。フォノン計算そのものには影響を与えません。
- lperm=.True.(for dynmat)
フォノン計算後のポスト処理(dynmat.x)においてΓ点の誘電率に対する寄与(イオン振動寄与分を含めた全誘電率)を算出します。
- ldisp=.true.
フォノン分散を計算します。フォノンバンド構造、フォノン状態密度を取得するためにはこれを指定する必要があります。
- nq1,nq2,nq3
フォノン分散の計算時のK点の数を指定します。
- Use spacing
Monkhorst-Pack法を使用します。各方向の分割数はSpacingパラメータ( Spacingパラメータ )で設定されます。
- NMR/EFGタブ
- Run GIPAW calculation (gipaw.x) as postprocess
GIPAW計算を実行します。 具体的には、pw.xを実行した後にgipaw.xを実行します。 gipaw.xの入出力ファイルは作業フォルダ内に作成されます。 GIPAW計算の結果の詳細を確認するには、gipaw.outまたはgipaw2.outを直接テキストエディタで開きます。
- job
GIPAW計算で出力する内容を指定します。「nmr & efg」を選んだ時はgipaw.xを2回起動し、それぞれjob=nmrとjob=efgで計算します。
- conv_threshold
対角化の打切り誤差を指定します。
- diagonalization
対角化方法を指定します。
- q_gipaw
線形応答理論で計算する際に使う、微小な波数の大きさを指定します。
- verbosity
ログへの出力レベルを指定します。
- use_nmr_macroscopic_shape/nmr_macroscopic_shape
サンプルのマクロな形状を考慮して化学シフトの値を補正するか指定します。
- spline_ps
擬ポテンシャルをスプライン補完するか指定します。
- q_efg
原子核の四重極モーメントを指定します。
- hfi_nuclear_g_factor
原子核のgテンソルを指定します。
- Enter q_efg
q_efgの入力補助機能です。デフォルトまたはユーザが用意したq_efgのテーブルから引用することができます。
- Open q_efg table
q_efgのテーブルを開きます。
- Dynamicsタブ
- Simulation Package
MD計算に使用する計算パッケージを指定します。cp.xの場合はCPMD法を使います。
- dt
MD計算の時間刻みをatomic unitで指定します。
- tempw
MD計算で温度制御を指定した際の目標温度を指定します。
- press
MD計算で圧力制御を指定した際の目標圧力を指定します。
- ion_temperature
MD計算のイオン(原子核)の温度制御方法を指定します。
- ion_velocities
MD計算のイオンの初速度を指定します。
- tolp
温度制御時の温度の許容値を指定します。
- pot_extrapolation
Born-Oppenheimer MD使用時のポテンシャルの外挿方法を指定します。
- wfc_extrapolation
Born-Oppenheimer MD使用時の波動関数の外挿方法を指定します。
- electron_dynamics
Car-Parrinello MD使用時のKS軌道を変化させるアルゴリズムを指定します。
- electron_velocities
Car-Parrinello MD使用時の電子の初速度を指定します。
- emass
Car-Parrinello MD使用時の電子の仮想質量を指定します。
- emass_cutoff
Car-Parrinello MD計算時の電子の仮想質量のカットオフ値を指定します。
- orthogonalization
行列計算(正規直交化)の方法を指定します。
- Dipole Corrタブ
- tefield
ノコギリ型の外部電場を印加します。
- dipfield
ダイポール補正を使用します。
- edir
tefieldとdipfieldが適用される方向を設定します。
- emaxpos
tefieldとdipfieldを適用する際の、外部電場が最大値となる場所を分率座標(0から1の範囲)で与えます。
- eopreg
tefieldとdipfieldを適用する際の、外部電場が減衰する領域のサイズを分率座標で与えます。
- eamp
tefieldとdipfieldを適用する際の、外部電場の大きさを与えます。
- ESMタブ
- assume_isolated=esm
ESM(Effective Screening Medium, 有効遮蔽媒質)法を使用する場合はチェックを入れます。
- esm_bc
ESM法で使われる境界条件の種類を指定します。
- esm_efield
電場を指定します。
- esm_w
ESMが設置される場所のオフセットを設定します。
- lfcpopt
化学ポテンシャル一定(constant mu)計算を実施します。初期の系全体の電荷はBasicタブのtot_chargeで指定されます。
- fcp_mu
化学ポテンシャル一定計算でのフェルミエネルギーの目標値を設定します。
- Enter Relative Potential
Target Fermi Energyの入力をサポートします。 まず、電圧0での計算のログファイルを指定し、電圧0でのフェルミエネルギーを取得します。 次に、印加電圧を入力します。 これら2つの情報から、Target Fermi Energyを算出します。
- RISM(1)タブ
- trism=.True.
RISM計算を有効にします。ESM-RISM計算を実行する際は、ここにチェックを入れ、 ESM タブの assume_isolated=esm にもチェックを入れてください。この機能を利用するには、ESM-RISM機能が有効になったバージョンのQuantum ESPRESSOを別途インストールする必要があります。
- closure
RISM計算で使用するクロージャを選択します。
- tempv
RISM計算の温度を指定します。
- ecutsolv
RISM計算のカットオフエネルギーを指定します。
- solute_lj
溶質(DFT領域)のLJパラメータを指定します。 none を選択した場合は、その下の solute_epsilon と solute_sigma にLJパラメータを入力してください。
- nsolv
溶媒の分子種の数を指定します。
- SOLVENTS
Density の単位をプルダウンから選択し、各溶媒分子種の密度(濃度)とMOLファイルの名前を指定します。MOLファイルは下の Directory for MOL Files で指定したフォルダに収められている必要があります。
- Directory for MOL Files
SOLVENTS で選択できるMOLファイルを収めたフォルダを指定します。
- RISM(2)タブ
- laue_expand_right/left
ESM-RISM計算における溶媒領域の遠方側の端の位置を指定します。
- laue_starting_right/left
ESM-RISM計算における溶媒領域の開始位置を指定します。
- laue_buffer_right/left
ESM-RISM計算における溶媒のバッファ領域の位置を指定します。
- Run only 1D-RISM
ここをチェックした場合は、pw.xの代わりに1drism.xを実行します。DFT計算は実行されません。溶媒原子間相関関数や溶媒間の化学ポテンシャルのみを知りたいときに有効です。
- rism3d_conv_level
SCF計算の各ステップにおける3D-RISM計算の打ち切り誤差を、動的に変化させる際のパラメータを指定します。
- rism1d/rism3d_maxstep
1Dおよび3D-RISMの最大反復回数を指定します。
- rism1d/rism3d_conv_thr
1Dおよび3D-RISMの打切り誤差を指定します。
- mdiis1d/3d_size
MDIISアルゴリズムによるRISM計算の収束パラメータを指定します。
- mdiis1d/3d_step
MDIISアルゴリズムによるRISM計算の収束パラメータを指定します。
- Othersタブ
QEの入力ファイルの書式(FORTRANのnamelist形式)で、その他の設定を記入します。 記入例は、ポインタを重ねると表示されます。
- Previewタブ
設定キーワードのプレビューが表示されます。
- Optionsタブ
- Verbosity
QEが出力する情報量を指定します。
- atomic_position unit
ATOMIC_POSITIONSおよびCELL_PARAMETERSの単位を指定します。
- Use max_seconds
チェックを入れた場合、ここに入力した秒数の経過後QEの処理が中断されます。
- Options for executable
pw.xやpw.exeを実行するコマンドの引数を与えます。例えば並列を制御する-nk, -nb, -nt, -ndなどを指定できます。
- Dump all files for remote
Linux環境でのジョブ実行に必要なファイルを出力します。リモートジョブ投入機能で生成されるファイルと同じファイルが出力されます。
- Open k-path file
ibrav(ブラべ格子)の種類ごとにデフォルトで指定するk点パスを記述する設定ファイル(UserPref/kpath_default.txt)を開きます。UserPref/kpath_default.txtが存在しない場合はwmx/kpath_default.txtからコピーされます。
- QE Version
出力するQEの入力ファイルが対応するQEのバージョンを指定します。いくつかのキーワード(&fcp, HUBBARDなど)の出力形式はバージョンによって変化します。
- Propertiesタブ
- Calculate these properties after pw.x
pw.x実行直後に実行されるポスト処理を選択します。ここで選択した処理の各種パラメータは右のParameter/Value欄にて指定します。
- Pseudopotentialタブ
- Mass
各元素の質量を指定します。
- Default
標準的な質量を設定します。
- Light
全元素の質量を1に設定します。イオンの構造緩和の促進などの目的で使われます。
- Manual
下のリスト内で、各元素に対し、ユーザーが個別に質量を設定します。
- Pseudopotential
系内の全元素に共通して存在する中から擬ポテンシャルファイルを選択します。
- (Type)
擬ポテンシャルの種類を選択します。
- (Functional)
汎関数の種類を選択します。(Type)で絞られた選択肢の中から選択します。
- (File)
擬ポテンシャルファイルを選択します。(Type)と(Functional)で絞られた選択肢の中から選択します。(Manual)を選んだ場合は、下のリスト内で、各元素に対し、ユーザーが個別に擬ポテンシャルを設定します。
擬ポテンシャルファイルは、pseudo Directoryで指定したフォルダの中から検索されます。 ユーザ設定フォルダの中の
qe_pseudo_priority_list.txtの先頭に書かれたものが優先的に選択されます。- Reload pseudo Files
pseudo Directoryで指定したフォルダに配置された擬ポテンシャルファイルを再度読み込みます。
- pseudo Directory
擬ポテンシャルファイルを配置するフォルダを指定します。 pseudo in QE's directory の場合は、QEのインストールディレクトリ以下のpseudoフォルダを用います。 (select...) の場合は、ダイアログで選択したディレクトリを用います。
- Open Pseudo Directory
pseudo Directoryで指定したフォルダを開きます。
- Download Pseudo Files
擬ポテンシャルファイルをダウンロードしてインストールします。
- Open Priority List
UserPref/qe_pseudo_priority.txtを開きます。存在しない場合はwmx/qe_pseudo_priority.txtからコピーされます。
- Reset
設定をリセットします。
- Import
設定ファイルを読み込みます。
- Export
設定ファイルを出力します。
6.16.3.1. Spacingパラメータ
Spacingパラメータは系のサイズによらず各方向のk点分割数
を決めるためのパラメータです。
(
は各方向
に対するインデックス)は格子ベクトル(基本並進ベクトル)
に対する逆格子ベクトル
です。
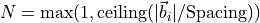
6.16.4. 実行
Quantum ESPRESSOを実行します。 状況に応じて実行方法が異なります。
CPMDを選択したときは cp.x 、それ以外の時は pw.x が実行されます。
(デフォルト) Output Directory = Create の時は、作業フォルダを新規に作成して計算を実行します。
Output Directory = Continue の時は、その時メインウィンドウで開いている入力ファイルのoutdirを作業フォルダとして使用します。
Output Directory = Select の時は、選択したフォルダを作業フォルダ(outdir)として使用します。
実行に伴い以下のファイルが生成されます。 例として入力ファイルが
si.pwinの時のファイル/フォルダ名を併記しています。
種類
説明
si.pwout計算のログファイルです。
si.batQuantum ESPRESSOを実行するためのバッチファイルです。
si_qe_data\作業フォルダには以下のファイルが生成されます。 ここでは主要なファイルのみ示しています。
種類
説明
*.UPFpw_bands.inpw_bands.outpw_bands.inのログファイルです。pw_dos.inpw_dos.outpw_dos.inのログファイルです。ph.inph.outph.inのログファイルです。ヒント
作業フォルダ
作業フォルダとは、メインウィンドウで開かれているファイルの名前に接尾辞を加えた名前のフォルダです。
接尾辞はソルバの種類により変わります。
例えばGromacsの場合は、メインウィンドウで開かれているファイルが
aaa.groで、接尾辞が_gmx_tmpの場合、作業フォルダの名前はaaa_gmx_tmpとなります。メインウィンドウで開かれているファイルと同じ階層に置かれている必要があります。
継続ジョブの時も同名の作業フォルダで処理が流れますが、デフォルトでは継続ジョブの実施直前に、直前のジョブの作業フォルダのバックアップが作成されます。
バックアップの名前は、重複する名前が存在しない範囲で一番小さい番号が付いたものになります。例えば、作業フォルダが
aaa_gmx_tmpのときはaaa_gmx_tmp1となります。番号が付いていないディレクトリが常に最新のものとなります。
ジョブは Winmostar Job Manager を通じて実行されます。
6.16.5. ログを表示
ログファイルをテキストエディタで開きます。
6.16.6. ログの抜粋を表示
ログファイルの主要な情報を抜粋して表示します。
6.16.7. アニメーション
6.16.7.1. 構造最適化/BOMD(pwout)
ログファイルの情報から構造最適化、BOMD計算等のアニメーションを作成し表示します。
CPMDの場合は CPMD(pos) を使用してください。
アニメーション表示の操作方法は アニメーション操作エリア を参照してください。 アニメーション操作エリアから動径分布関数、自己拡散係数、平均二乗変位、各原子の変位などを計算できます。
6.16.7.2. CPMD(pos)
CPMDのposファイルとcelファイルを指定し、アニメーションを表示します。
pw.xの結果を表示する場合は 構造最適化/BOMD(pwout) を使用してください。
アニメーション表示の操作方法は アニメーション操作エリア を参照してください。 アニメーション操作エリアから動径分布関数、自己拡散係数、平均二乗変位、各原子の変位などを計算できます。
6.16.8. エネルギー変化
6.16.8.1. SCFエネルギー変化(pwout)
ログファイルを選択し、total energyのグラフを表示します。
CPMDの場合は CPMDエネルギー変化(evp) を使用してください。
- Property
グラフ表示する値を選択します。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.8.2. CPMDエネルギー変化(evp)
CPMDのevpファイルを指定し、各種エネルギーの時間変化を表示します。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9. 結果解析
6.16.9.1. 状態密度
作業フォルダ(outdir)とSCF計算のログファイルを指定し、状態密度を表示します。
SCF計算のログファイルからはフェルミエネルギーを取得します。 バックグラウンドでdos.xが流れます。
フェルミエネルギー付近の状態密度が0となるエネルギーの範囲が内部的に計算され、バンドギャップ、VBM(価電子帯上端)、CBM(伝導帯下端)のエネルギーの値が算出され表示されます。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.2. 部分状態密度
作業フォルダ(outdir)とSCF計算のログファイルを指定し、部分状態密度(PDOS)を表示します。
SCF計算のログからはフェルミエネルギーを取得します。 バックグラウンドでprojwfc.xが流れます。
- Check atom using viewport
ログファイルから読み込まれた分子構造が3D表示されます。そこでクリックして選択された原子の軌道が、軌道のリストの中でチェックされます。
- Show band center
各軌道のバンド中心を計算します。dバンド中心などを取得することができます。積分範囲に伝導帯を含めるか否かを選択できます。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.3. バンド構造
作業フォルダ(outdir)とSCF計算のログファイルを指定し、バンド構造を表示します。
事前にcalculation=bandsで計算が終了している必要があります。 SCF計算のログファイルからはフェルミエネルギーを取得します。 バックグラウンドでbands.xが流れます。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.4. Lowdin電荷
作業フォルダ(outdir)を指定し、Lowdin電荷を算出・表示します。
バックグラウンドでprojwfc.xが流れます。
6.16.9.5. Lowdin電荷
作業フォルダ(outdir)を指定し、Bader電荷を算出・表示します。あらかじめPAW法で全電子密度のcubeファイルを生成しておく必要があります。
バックグラウンドでbaderプログラムが流れます。
6.16.9.6. 電子密度
作業フォルダ(outdir)を指定し、等電子密度面を表示します。
バックグラウンドではpp.xが流れ、cubeファイルが生成されます。
サブウィンドウの操作方法は Surface Setup・Cubegenウィンドウ を参照してください。
6.16.9.7. スピン密度
作業フォルダ(outdir)を指定し、等スピン密度面を表示します。
バックグラウンドではpp.xが流れ、cubeファイルが生成されます。
サブウィンドウの操作方法は Surface Setup・Cubegenウィンドウ を参照してください。
6.16.9.8. ポテンシャルエネルギー
作業フォルダ(outdir)を指定し、等ポテンシャルエネルギー面を表示します。
バックグラウンドではpp.xが流れ、cubeファイルが生成されます。
サブウィンドウの操作方法は Surface Setup・Cubegenウィンドウ を参照してください。
6.16.9.9. ポテンシャルエネルギー分布
作業フォルダ(outdir)とSCF計算のログファイルを指定し、z軸方向のポテンシャルエネルギー分布(交換相関エネルギーを除く)を表示します。
SCF計算のログからはフェルミエネルギーを取得します。 フェルミエネルギーとポテンシャルエネルギー分布(交換相関エネルギーを除く)の最大値の差を、仕事関数の推定値として表示します。 バックグラウンドでpp.xとaverage.xが流れます。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.10. フェルミ面
SCF計算とbands計算のログファイルを指定し、フェルミ面を表示します。
フェルミ面の表示には FermiSurfer を使用します。 # of K Points にbands計算時のk点分割数を指定し、 Calc ボタンを押すとフェルミ面が表示されます。
6.16.9.11. 誘電関数
誘電関数計算後の作業フォルダを指定し、誘電関数を表示します。
- Direction
取得する誘電関数の方向を指定します。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.12. NMR
GIPAW計算後の作業フォルダを指定し、NMRスペクトルを表示します。
サブウィンドウの操作方法は NMRウィンドウ を参照してください。
6.16.9.13. IR/ラマンスペクトル
フォノン計算後の作業フォルダとSCF計算のログを指定し、IRおよびラマンスペクトルを表示します。
サブウィンドウの操作方法は IR Spectrumウィンドウ を参照してください。
6.16.9.14. フォノン分散曲線
フォノン分散計算後の作業フォルダを指定し、フォノン分散曲線を表示します。
- ASR
適用するAcoustic Sum Ruleの種類を指定します。
- K Points
取得する分散曲線のパスを指定します。 各行には、x, y, z成分をそれぞれ2pi/aの単位で記述し、その横に次の点までの間の分割数を記述します。(合計4カラムをスペース区切りで入力)
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.15. フォノン状態密度
フォノン分散計算後の作業フォルダを指定し、フォノン状態密度を表示します。
- ASR
適用するAcoustic Sum Ruleの種類を指定します。
- K Points
フォノンDOS計算時のK点の分割数を指定します。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.16. 電荷/エネルギー分布 (esm1)
ESM計算(assume_isolated=esm)で出力されるesm1ファイルを指定し、z軸方向の電荷またはエネルギーの分布を表示します。
2つのesm1ファイルの差をプロットすることもできます。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.17. 1D-RISM RMS変化 (pwout)
RISM計算(trism=.True.)の最初に実行される1D-RISM計算のRMSの変化をプロットします。
- Draw
グラフを表示します。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.18. 3D/Laue-RISM RMS変化 (pwout)
RISM計算(trism=.True.)実行時の、最後のSCFステップにおける3D-RISMまたはESM-RISM計算のRMSの変化をプロットします。
- Draw
グラフを表示します。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.19. 溶媒原子間相関関数 (1drism)
RISM計算(trism=.True.)で出力される1drismファイルを用いてRISM領域の原子間相関関数(動径分布関数)を算出します。
- Obtain Chemical Potentials
溶媒分子間の化学ポテンシャルをcsv形式で出力します。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.9.20. 溶媒密度/エネルギー分布(rism1)
RISM計算(trism=.True.)で出力されるrism1ファイルを用いて、DFT領域-RISM領域が接する方向(界面垂直方向)の溶媒密度・エネルギー・電荷を算出します。
- Draw
グラフを表示します。 必要に応じて結果解析用プログラムが実行されます。
- Close
ウィンドウを閉じます。
グラフ描画エリアの操作方法は グラフの操作方法 を参照してください。
6.16.10. MOLファイルを作成
RISM計算(trism=.True.)で使用される溶媒のMOLファイルを作成します。溶媒分子を1分子メインウィンドウに作成した上で本機能を呼び出してください。LAMMPSのdata形式のファイルからLJパラメータを設定したい場合は、data形式のファイルをメインウィンドウで開いた上で Use parameters in displayed file にチェックを入れてください。
作成したファイルはMOLファイルのキーワード設定ウィンドウの RISM(1) タブの Directory for MOL Files で指定したフォルダに配置してください。
6.16.11. Nudged Elastic Band
6.16.11.1. ワークフロー設定/キーワード設定
NEB計算の条件を設定し実行します。NEBの実行には
neb.xを使います。始状態と終状態それぞれの構造最適化計算を終えた状態で設定してください。
6.16.12. 実行
ファイルモードでQuantum ESPRESSOのNEB計算を実行します。
6.16.12.1. ログを表示
NEB計算のログファイルをテキストエディタで開きます。
6.16.12.2. NEBエラー変化
NEB計算の各iterationにおけるエラーを表示します。
6.16.12.3. アニメーション
NEB計算の出力ファイルを指定し、NEB計算収束後の後の反応座標に沿ったエネルギーと原子構造の変化を表示します。NEB計算の各iterationにおけるエネルギーの変化を連続的に確認したい場合は エネルギー分布 を利用してください。
アニメーション表示の操作方法は アニメーション操作エリア を参照してください。
6.16.12.4. エネルギー分布
NEB計算の出力ファイルを指定し、反応座標に沿ったエネルギーの変化を表示します。NEB計算の各iterationにおける様子を可視化できます。原子構造を可視化したい場合は アニメーション を使用してください。
6.16.13. BoltzTraP
BoltzTraPでは、QuantumESPRESSOでのnscf計算のアウトプットをもとに熱電特性の計算を行います。
6.16.13.1. キーワード設定・実行
BoltzTraPを用いたプリ・ポスト処理の計算条件を設定し、プリ処理の実行を行います。
Create .intransファイル ボタン
Quantum ESPRESSOのnscf計算のアウトプットファイル (.pwout)を読み込み、BoltzTraP設定ファイル(.intrans)を生成します。 アウトプットファイルを仮にmg2si_nscf.pwoutとすると、同じ階層にmg2si_nscfという名前の作業フォルダを生成します。 mg2si_nscf.intransはmg2si_nscf内に生成されます。
intransファイルの作成に成功した場合、ファイルの内容を読み込み、下記キーワードの入力欄に反映します。
- Fermilevel (Ry)
pwoutファイルから読み込んだフェルミエネルギーが設定されています。
- energy grid
設定エネルギーの間隔を指定します。
- energy span
フェルミレベル周辺の計算に考慮に入れるバンドエネルギーの範囲を指定します。
- number of electrons
単位格子内の電子の個数を指定します。
- lpfac
バンドエネルギーをフーリエ展開によって補完する際の因子を指定します。
- efcut
化学ポテンシャルを変えて計算する範囲を指定します。
- Tmax
設定温度の上限値を指定します。
- temperature grid
設定温度の間隔を指定します。
- energy of bands
DOSから得られたバンドのエネルギー幅を指定します。
- Calculate expansion coeff
チェックが入っている場合、展開係数を計算します(CALC)。入っていない場合、展開係数をファイルから読み込みます(NOCALC)。
Start BoltzTraP ボタン
設定条件を元にintransファイルを再作成し、BoltzTraPを実行します。 このとき、以下のファイルとフォルダが生成されます。
下記では、計算終了時点での作業フォルダ(mg2si_nscf)内の主なファイルを記載します。
種類
説明
mg2si_nscf.intransBoltzTraPのインプットファイルです。
mg2si_nscf.traceBoltzTraPで計算された熱電特性のエネルギー、温度依存性に関する情報が出力されたファイルです。 BoltzTraP結果読み込みメニューではこのファイルを読み込み可視化を行います。
Cancel ボタン
何もせずにキーワード設定・実行ウィンドウを閉じます。
6.16.13.2. 結果読み込み
BoltzTraPによって計算された下記の熱電特性を読み込み、可視化を行います。
ゼーベック係数(Seebek coefficient)
電気伝導度(Electrical conductivity)
熱伝導率(Electrical thermal conductivity)
出力因子(Power factor)
性能指数(Figure of merit)
各温度における特性値のエネルギー依存性を表示したい場合はコンボボックスからT [K]を選択し、リストから目的の温度を選択します。 各エネルギーにおける特性値の温度依存性を表示したい場合はコンボボックスからE-Ef [eV]を選択し、リストから目的のエネルギーを選択します。
6.16.14. Phonopy
Phonopyを用いた計算を以下の3つの工程で計算を行います。
与えられた結晶構造(プロジェクトモード)またはQuantum ESPRESSOインプットファイル(ファイルモード)を元にスーパーセルを作成する。
生成された全てのスーパーセルに対し、Quantum ESPRESSOを実行する。
Quantum ESPRESSOのアウトプットファイルから、ForceSetsファイルを作成し、 Phononバンド、DOS、熱物性などの計算を行う。
6.16.14.1. スーパーセルを作成
プロジェクトモードにおいて、分子表示エリアに表示されている結晶構造に対しPhonopy計算用のスーパーセルを生成します。結晶の種類によって複数の構造が生成されることがあります。それらの構造に対しQuantum ESPRESSOで計算をするには、 ワークフロー設定 でEnable scan calculationを使用します。
6.16.14.2. キーワード設定・実行
ファイルモードにおいて、Phonopyを用いたプリ・ポスト処理の計算条件を設定し、プリ処理の実行を行います。
- Open ボタン
Quantum ESPRESSOのインプットファイル (*.in,*.pwin)を読み込みます。 Phonopyでのポスト処理では、応力の情報を必要とします。このため読み込むファイルは tprnfor, tstressキーワードを含んでいる必要があります。 Phonopy用のQuantum ESPRESSOのインプットファイルは、 Quantum ESPRESSOキーワード設定のPreset=Phonopyを使用することで設定できます。
- DIM
スーパーセルのx, y, z方向のリピート回数を半角スペース区切りで指定します。
- MP
PhonopyでPhonon DOSや熱物性の計算を行う際の逆格子を半角スペース区切りで指定します。
- ATOM_NAME
単位格子に含まれる元素を半角スペース区切りで指定します。 Open ボタンでインプットファイルを開いた時点で自動的に入力されます。
Start ボタン
設定条件を元にPhonopyを実行し、プリ処理であるスーパーセルの作成を実行します。 このとき、以下のファイルとフォルダが生成されます。
種類
説明
si.batPhonopyのプリ処理を実行するためのバッチファイルです。
si.shPhonopyのプリ処理を実行するためのシェルスクリプトファイルです。
si_ph_data計算の作業フォルダです。
作業フォルダsi_ph_dataには以下のファイルが生成されます。
種類
説明
mesh.confPhonopyのポスト処理にて、状態密度や熱物性を計算するときに使用されます。
band.confPhonopyのポスト処理にて、バンド構造を計算するときに使用されます。
header.insupercell-*.intmp-*.inCancel ボタン
何もせずにキーワード設定・実行ウィンドウを閉じます。
6.16.14.3. confファイル編集
ファイルモードにおいて、confファイルをテキストエディタで開きます。 キーワード設定画面で設定したキーワードを編集したい場合に用います。
6.16.14.4. Quantum ESPRESSO連続実行
ファイルモードにおいて、キーワード設定・実行画面で生成された、全てのスーパーセルに対し、Quantum ESPRESSOを実行します。 このメニューを用いた場合、Quantum ESPRESSOはローカル環境で実行されます。
6.16.14.5. Phonopy実行
ファイルモードにおいて、Phonopyのポスト処理を実行します。
この時、作業フォルダ :file: si_ph_data に以下のファイルが生成されます。
種類
説明
phonopy.shPhonopyのポスト処理を実行するためのシェルスクリプトです
band.yamlPhonopyのポスト処理で計算されたバンド構造の情報が出力されています。
dos.datPhonopyのポスト処理で計算された状態密度の情報が出力されています。
thermal_properties.yamlPhonopyのポスト処理で計算された熱物性の情報が出力されています。
6.16.14.6. バンド構造
作業フォルダに含まれるband.csvをもとにバンド構造を表示します。
6.16.14.7. 状態密度
作業フォルダに含まれるtoal_dos.csvをもとに状態密度を表示します。
6.16.14.8. 熱物性
作業フォルダに含まれるthermal_properties.csvをもとに熱物性を表示します。
6.16.15. 弾性定数テンソル
弾性定数テンソルを計算します。あらかじめ 構造スキャン 機能でGenerate all strain modesに設定して生成した構造に対し、構造スキャン計算を実行しておく必要があります。