6.3. 選択メニュー
原子・分子を選択する機能に関するメニューです。
ヒント
原子の選択方法
原子を選択する方法には、 赤丸のマーカーを用いる方法 と、 青丸のグループ選択を用いる方法 があります。
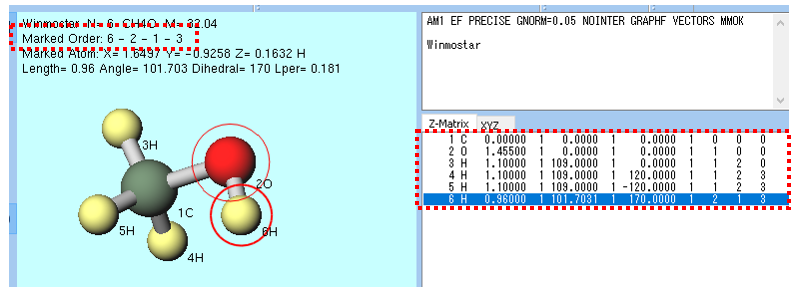
赤丸のマーカーを用いる方法 は主に1原子に対する操作で使われます。分子表示エリアで 左クリック した原子にマーカーが移動します。下図のメタノール分子の例では、OH(ヒドロキシ)基の酸素(赤)→水素(黄色)の順に 左クリック しており、最後にマーカーが付けられた原子に太赤丸、1つ前にマーカーを付けられた原子に細赤丸が付いています。また、下図右赤枠内の、 座標表示エリア で 左クリック することでもマーカーが移動します。マーカーが付けられた原子は過去4つ分記憶され、下図左上赤枠内のように表示されます(下図の場合は Marked Order: 6 - 2 - 1 - 3 )。各原子の番号は 座標表示エリア または を選択することで 分子表示エリア 内で確認できます。
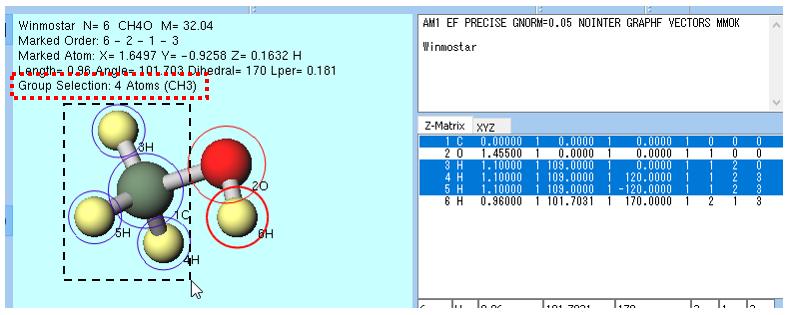
青丸のグループ選択を用いる方法 は、主に複数原子に対する操作で使われます。 分子表示エリア で Ctrl+左ドラッグ 、 Ctrl+左クリック 、 Shift+左クリック することでグループ選択できます。あるいは メニュー以下の機能を使うことでもグループ選択できます。下図のメタノール分子の例では、CH3(メチル)基の周囲を Ctrl+左ドラッグ しており、CH3基の原子が青丸で囲まれています。また、 座標表示エリア で Ctrl+左クリック することでもグループ選択できます。 分子表示エリア 内の数左上にはグループ選択されている原子の個数と組成が表示されます(下図の例では Group Selection: 4 Atoms (CH3) )。
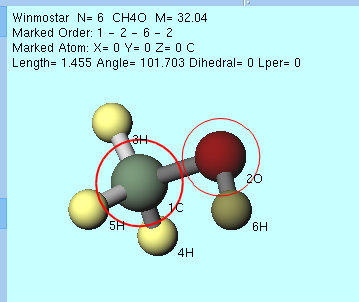
グループ選択されていない状態で、 などの複数原子に対する操作を実行した場合は、 マーカーで分断される部分構造をグループ選択 が自動で実行されます。具体的には、赤太丸と赤細丸のマーカーが付けられた2原子で分断される部分構造がグループ選択されます。下図のメタノール分子の例では、酸素(赤)、炭素(緑)と順に 左クリック してマーカーを移動させた後、 グループを軸回転(選択2原子,マウス操作) を選択すると、酸素-炭素の間で分断される構造のうち、最後にマーカーが付けられたCH3基の側が自動でグループ選択されます(画面上ではハイライトされる)。
6.3.1. すべてをグループ選択
全ての原子をグループ選択します。
6.3.2. グループ選択を解除
グループ選択を解除します。
6.3.3. グループ選択の範囲を反転
グループ選択されていない原子をグループ選択し、それまでグループ選択されていた原子のグループ選択は解除します。
6.3.4. 分子種によるグループ選択
Select by ウィンドウの Use List タブが Molecular Species にチェックされた状態で開きます。 Select by ウィンドウ内のリストの各行をクリックすると、対応する分子種がグループ選択されます。
注釈
タンパク質のpdbファイルから計算する際に本機能を使用するとタンパク・リガンド・結合水・緩衝剤などを抜き出すことができます。
6.3.5. 分子によるグループ選択
Select by ウィンドウの Use List タブが Molecules にチェックされた状態で開きます。 Select by ウィンドウ内のリストの各行をクリックすると、対応する分子がグループ選択されます。
6.3.6. 元素によるグループ選択
Select by ウィンドウの Use List タブが Elements にチェックされた状態で開きます。 Select by ウィンドウ内のリストの各行をクリックすると、対応する元素がグループ選択されます。
6.3.7. 選択記述言語によるグループ選択
Select by ウィンドウの Use Selection Language タブが開きます。 テキストボックスに選択記述言語で選択方法を記述した後に、 Apply ボタンをクリックすると、対応する原子がグループ選択されます。 選択記述言語では以下の構文がサポートされています。
基礎的な構文
element C H全ての炭素と水素原子を選択します
index 1-3 6 81, 2, 3, 6, 8番目の原子を選択します。
compid 1
moleid 1-3
site 1
charge 0.5
resname GLY残基名がGLYの残基に含まれる原子を選択します。
name CA原子名がCAの原子を選択します。
x< 10x座標が10 Angstrom未満の座標を選択します。(xと<の間にスペースなし)同様にy,zについても使用可能です。
x>,x=>,x<=も使用可能です。
bondto HH原子に結合する原子を選択します。
group mygroupmygroupという登録グループを選択します。
残基名、原子名はPDBまたはgro形式のファイルを開いた際に使用可能です。
論理演算子を用いた複合的な構文
(compid 1) and (site 1)CompIDが1の分子種で、かつ分子内の1番目のサイトを選択します。
(current) and (element H)現在グループ選択されている原子の中で水素原子だけを選択します。
(resname GLY) and (not (element H))残基名がGLYで、かつ水素以外の原子を選択します。
and、not以外にorとxorを使用可能です。 これらの論理演算子を使用する際には上記の例のように丸括弧()を使用してください。また、整数を入力するところに
%2==0と入力すると2で割った余りが0になる数列が代入されます。例えばsite %3==1と入力するとサイトが1, 4, 7, 10...の原子が選択されます。
6.3.8. マーカーで分断される部分構造をグループ選択
1番目と2番目のマーカーが付いた2個の原子の間で分断される部分構造をグループ選択されます。 詳細は 選択メニュー に画像付きで記載しています。
6.3.9. 現在のグループに結合する原子をグループ選択
現在の選択グループに結合する原子を追加でグループ選択します。
6.3.10. 現在のグループに隣接する原子をグループ選択
現在の選択グループに隣接する原子をグループ選択します。ここでは、全原子にボロノイ分割を適用した際に、同一のボロノイ境界を有する原子を、隣接する原子を定義しています。
6.3.11. 現在のグループに隣接する分子をグループ選択
現在の選択グループに隣接する分子をグループ選択します。ここでは、全原子にボロノイ分割を適用した際に、同一のボロノイ境界を有する原子を、隣接する原子を定義しています。
6.3.12. マークした原子の周辺の原子をグループ選択
マークした原子から指定距離内に存在する原子をグループ選択します。
6.3.13. マークした原子の周辺の分子をグループ選択
マークした原子から指定距離内に重心が存在する分子をグループ選択します。
6.3.14. マークした原子を含む残基をグループ選択
マークした原子を含む残基をグループ選択します。残基番号が同じで原子のIDが連続しているものが同じ残基であると判定されます。
6.3.15. 分子種ごとに拡大表示してグループ選択
分子種ごとに分子を3Dで拡大表示して、原子をグループ選択できるようにします。Create Groupウィンドウで、Targetで分子種を切り替え、ウィンドウ左側でCtrl+クリックで原子(サイト)を選択します。Addボタンをクリックすると選択されたサイトが登録され、OKをクリックすると、各分子の登録されたサイトについてのグループが作成されます。Apply to First Molecule Onlyにチェックを入れた場合は、選択した分子種の1番目の分子に対してのみ処理を実行します。
6.3.16. グループを登録
グループ選択されたグループに名前を付けて登録し、 登録グループを選択 から呼び出せるようにします。
6.3.17. 登録グループを選択
グループを登録 で登録されたグループを呼び出します。LAMMPS実行時には、ここにリストアップされたグループがndxファイル内に出力されます。Winmostarを再起動した後も登録グループを引き継ぎたい場合は、 登録グループをndxファイルに書き出し を用いてndxファイルに書き出した後、 ndxファイルからグループを読み込み を用いて読み込みます。
6.3.17.1. 登録されたグループの名前を変更
グループを登録 で登録されたグループの名前を変更します。
6.3.17.2. 登録されたグループを削除
グループを登録 で登録されたグループを削除します。
6.3.18. ndxファイルからグループを読み込み
ndxファイルに出力されたグループを読み込み、 登録グループを選択 から呼び出せるようにします。
6.3.19. 登録グループをndxファイルに書き出し
登録グループを選択 に登録されたグループをndxファイルに出力します。
6.3.20. グループをndxファイルに追加
グループ選択されたグループを、指定したndxファイルに追加します。
6.3.21. グループを結合して転置
含まれる原子の数が等しい複数のグループを結合し、順序を転置します。例えば、グループAに1 2 3、グループBに4 5 6が登録されていて本機能をグループAとグループBに適用した場合、1 4、2 5、3 6という3つのグループが作成されます。